Claudia Sorrentino1, Antonio Cuneo1,2 and Giovanni Roti1.
1 University of Parma, Department of Medicine and Surgery, Parma, 43126, Italy.
2 University of Ferrara, Department of Medical Sciences, Ferrara, 44121, Italy.
Correspondence to: Giovanni Roti. University of Parma, Department of Medicine and Surgery, Parma, 43126, Italy. E-mail:
giovanni.roti@unipr.it
Published: July 1, 2019
Received: March 3, 2019
Accepted: May 18, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019037 DOI
10.4084/MJHID.2019.037
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
Notch pathway plays a key role in several processes, including
stem-cell self-renewal, proliferation, and cell differentiation.
Several studies identified recurrent mutations in hematological
malignancies making Notch one of the most desirable targets in leukemia
and lymphoma. The Notch signaling mediates resistance to therapy and
controls cancer stem cells supporting the development of on-target
therapeutic strategies to improve patients’ outcome. In this brief
review, we outline the therapeutic potential of targeting Notch pathway
in T-cell acute lymphoblastic leukemia, chronic lymphocytic leukemia,
and mantle cell lymphoma..
|
Introduction
Notch
pathway comprises a family of single-pass transmembrane receptors,
their ligands, and coactivators that regulate evolutionarily conserved
signaling that controls development and tissue homeostasis[1,2]
in all metazoan organisms. Mammalian NOTCH receptors (NOTCH 1-4) are
pre-processed during maturation by a furin-like protease (S1), leading
to the formation of two, non-covalently associated subunits. In
non-malignant cells, canonical Notch signaling is initiated by
cell-to-cell contact of the Notch extracellular domain (NECD) to a
ligand of the Delta-like (DLL1, DLL3, DLL4) and Jagged family (JAG1,
JAG2), expressed on the cellular surface of the neighboring cell. This
receptor-ligand interaction mediates a sequence of two proteolytic
cleavages in the Notch transmembrane subunit. The first, resolved by
ADAM-10 or ADAM-17 metalloproteases, occurs within a juxtamembrane
negative regulatory region (NRR) at a site that is protected in the
inactive state (S2).[3-5] This cleavage generates a trans-membrane intermediate that is the substrate for a secondary cleavage (S3) by the γ-secretase, an event that releases the intracellular domain of NOTCH (ICN, NICD).[6]
ICN moves to the nucleus, complexes with the DNA-binding factor RBPJ,
and recruits coactivator of the Mastermind-like (MAML) family. The
resulting macromolecules complex activates genes transcription but is
usually short-lived because the C-terminal portion of ICN (PEST,
peptide sequence that is rich in proline (P), glutamic acid (E), serine
(S), and threonine (T)) is recognized by an E3 ubiquitin ligase and
degraded.[7]
The NOTCH proteins have several
functional domains organized in modules. The NECD N- terminal domain is
responsible for ligand binding through EGF-like Ca2+ dependent repeats,
followed by three LNR (Lin12/Notch) units. Next to the LNR region lays
the juxtamembrane heterodimerization domain (HD), a linker between the
extracellular tail and ICN. LNR and HD modules constitute the negative
regulatory region (NRR) that prevents ADAM-10/17 cleavage of mammalian
Notch in the ligand's absence (Figure 1A).[3-5,8]
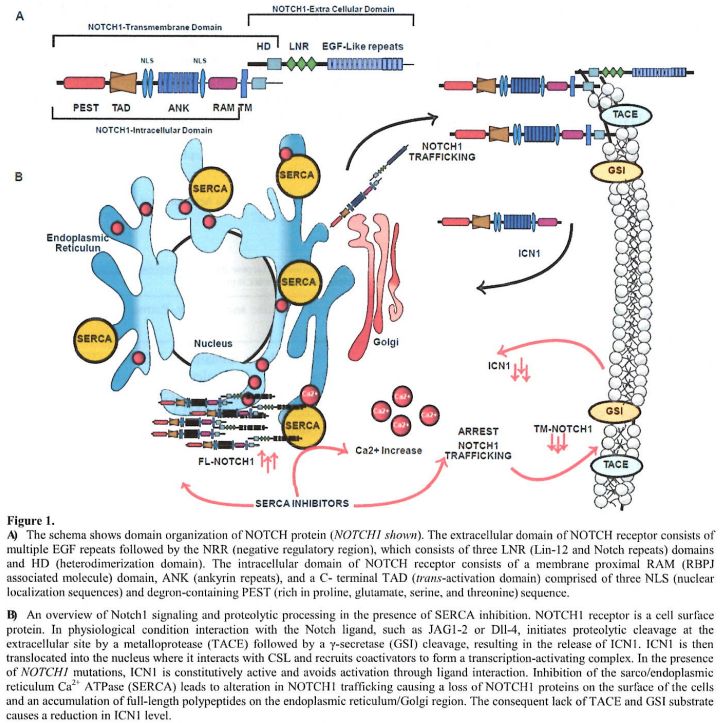 |
Figure
1. A) The
schema shows domain organization of NOTCH protein (NOTCH1 shown). The
extracellular domain of NOTCH receptor consists of multiple EGF repeats
followed by the NRR (negative regulatory region), which consists of
three LNR (Lin-12 and Notch repeats) domains and HD (heterodimerization
domain). The intracellular domain of NOTCH receptor consists of a
membrane proximal RAM (RBPJ associated molecule) domain, ANK (ankyrin
repeats), and a C- terminal TAD (trans-activation domain) comprised of
three NLS (nuclear localization sequences) and degron-containing PEST
(rich in proline, glutamate, serine, and threonine) sequence.
B) An
overview of Notch1 signaling and proteolytic processing in the presence
of SERCA inhibition. NOTCH1 receptor is a cell surface protein. In
physiological condition interaction with the Notch ligand, such as
JAG1-2 or Dll-4, initiates proteolytic cleavage at the extracellular
site
by a metalloprotease (TACE) followed by a γ-secretase (GSI) cleavage,
resulting in the release of ICN1. ICN1 is then translocated into the
nucleus where it interacts with CSL and recruits coactivators to form a
transcription-activating complex. In the presence of NOTCH1 mutations,
ICN1 is constitutively active and avoids activation through ligand
interaction. Inhibition of the sarco/endoplasmic reticulum Ca2+ ATPase
(SERCA) leads to alteration in NOTCH1 trafficking causing a loss of
NOTCH1 proteins on the surface of the cells and an accumulation of
full-length polypeptides on the endoplasmic reticulum/Golgi region. The
consequent lack of TACE and GSI substrate causes a reduction in ICN1
level. |
While oncogenic alterations in the Notch signaling have been described in almost all human cancers,[3,9] the majority of the recurrent somatic mutations of NOTCH proteins are observed in the NOTCH1 gene.
The role of NOTCH1 in the pathogenesis of T-cell acute lymphoblastic leukemia (T-ALL), was first investigated in 1991.[1]
Ellisen and colleagues described a chromosome translocation,
t(7;9)(q34;q34), that juxtaposes the T-cell receptor-β to the active
form of ICN1 in T-ALL.[10] This fusion creates
an oncogenic Notch1 signaling in leukemia cells. Similarly, to the
translocation, activating NOTCH1 mutations generate ligand independent
or proteasome resistant ICN1 peptides that sustain T-cell
transformation, leukemia growth, or resistance to therapy.[10] In T-ALL, NOTCH1 mutations cluster in two different but not mutually exclusive hotspots.[11,12]
The first comprises a single amino acid substitution and in-frame
insertion in the extracellular NRR. To this class also belongs the rare
in-frame insertion in the juxtamembrane extracellular domain (JME).
Within the NRR module, most of these mutations occur in the HD domain,
and they are defined as type 1A and 1B.[13] Briefly,
HD mutations cause ligand-independent Notch conformational changes that
constitutively activate ICN1. The second hotspot of NOTCH1 mutations
comprises small insertion/deletion in the exon 34 (PEST domain). These
genetic lesions truncate NOTCH1 C-terminal generating a long-lived ICN1
caused by the consequent loss of the “degron” recognition site of the
PEST unit.[11,14]
Recently,
NOTCH1 emerged as one of the most frequently mutated genes (~5-20%) in
chronic lymphocytic leukemia (CLL), where it may represent an early
driver lesion in a proportion of cases.[15,16] Most
of these mutations, ~80%, are a 2-bp deletion in exon 34 that generates
a premature stop codon (P2514fs*4), that truncates the PEST region.
Similarly to T-ALL, these mutations cause an over-activation of Notch1
signaling because of the lack of its degradation.[17]
Interestingly Kridel and colleagues reported a similar pattern of
mutations within the PEST domain in mantle cell lymphoma (MCL).[18,19]
Furthermore, 50% of NOTCH1 wild-type CLL cases express ICN1 suggesting
that the activation through the canonical Notch signaling is required
for leukemia growth in this disease.[20] However, in CLL and MCL, mutations in NOTCH1 are associated with a worse prognosis.[17,21-23]
In addition to these observations, Schmitz and colleagues recently
described a genetic framework for diffuse large B-cell lymphoma (DLBCL)
that may influence the therapeutic response.[24] They
identified gain-of-function NOTCH1 mutations (“N1”; these mutations
mainly occur in the PEST region) in 19/574 cases of DLBCL. Among these
cases, 95% were activated B-cell-like (ABC) diffuse large B-cell
lymphoma and no other type of mutation (BCL6 fusions (B) NOTCH2 (N2),
or SPEN mutations) co-occurred suggesting that NOTCH1 and NOTCH2 act
through different pathogenetic pathways.[24]
Moreover, within ABC DLBCL, patients with N1 mutation had worse
progression-free survival and overall survival compared to patients
with N2 mutation.[24] These data highlight that N1
and N2 mutations are genetically, phenotypically, and clinically
different, suggesting the need to extend targeting Notch1 in these
aggressive forms of B-cell malignancies.
Here we review some of
the latest strategies to target Notch in hematological malignancies
with emphasizing innovative approaches or experiences that translated
pre-clinical observations into clinical trials (Figure 2).
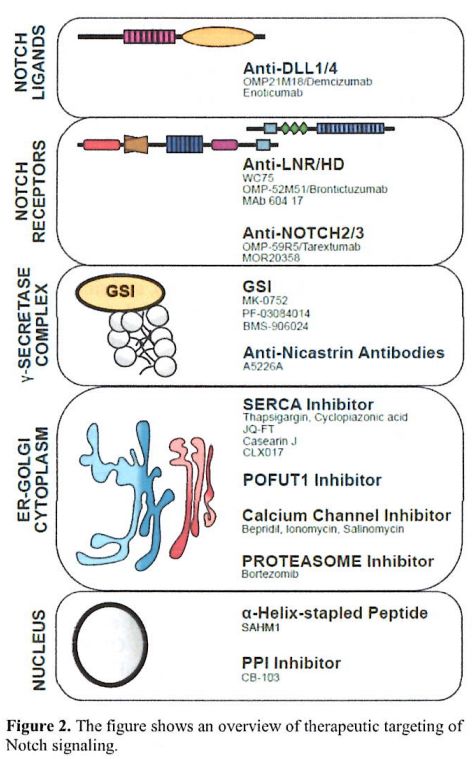 |
Figure 2. The figure shows an overview of therapeutic targeting of Notch signaling. |
Targeting Extracellular NOTCH1
Unlike
Notch pathway activation in mutated T-ALL, CLL, MCL, the canonical
activation of Notch signaling is mediated by ligand-mediated
mechanisms.[25,26] Thus, given the role of Notch in
several humans’ cancers, the development of therapeutic agents that
interfere with ligand-receptor binding has seen a great impetus in the
last years.[27]
A strategy that has been
extensively explored is the development of antibodies (Abs) to block
Notch ligand-receptor interaction. Several groups developed
receptors-directed antibodies designed to antagonize NOTCH1, 2 and 3 by
recognizing the NRR region of NOTCH to prevent the ADAM mediated
metalloprotease cleavage.[28-30]
For example,
Aste-Amezaga reported the identification of two classes of NOTCH1
inhibitory monoclonal (m) Ab derived from cell-based and solid phase
screening of a phage display library.[31] The first
class comprises Abs directed to the EGF-repeat region (WC613), and the
second directed to the NRR NOTCH1 domain (WC75). Both classes of
antibodies inhibited canonical Notch signaling in vitro by repressing
Notch transcriptional targets such as Hes1 and DTX1 genes. As predicted
by the analysis of the putative NOTCH1 binding site, WC75 also
inhibited Notch activation in a ligand-independent fashion such as in
cancers mutated models (T-ALL), and similar to a γ-secretase
inhibitor, Compound E, induced a gene expression signature consistent
with Notch1 abrogation. Consistently WC75 inhibited the proliferation
of NOTCH1 mutated T-ALL cell lines such as DND41 and KOPT-K1.[31]
Similarly
OMP-52M51, a mAb generated by immunizing mice with a fragment of human
NOTCH1 protein comprising the LNR plus the HD domain, efficiently
blocked canonical Notch signal and reduced Notch activation in a series
of T-ALL bearing HD and PEST mutations in vitro and in two
patient-derived xenograft leukemia models carrying a L1679P mutation
and a PEST deletion respectively.[32] In addition, OMP-52M51 prevented Notch1 activation in MCL cell lines in vitro.[33]
OMP-52M51 (Brontictuzumab) was subsequently tested in a phase I dose
escalation trial (NCT01778439) in patients with previously treated CLL,
MCL, T-ALL, or other hematologic malignancies with known NOTCH1
mutational status. Of the 24 patients enrolled in this study, only five
carried a NOTCH1 mutation, and just one of them achieved stable disease
as the best response after 101 days of treatment. Overall OMP-52M51 was
generally well tolerated but showed limited antitumor efficacy in this
study.[34]
However, Sharma and colleagues
further extended targeting NRR domain and reported the identification
of the first mAb that recognizes clinically relevant mutant receptors.[35]
The mAb 604.17 exhibited higher binding to mutant NOTCH1 compared to
wild type and inhibited the proliferation of the T-ALL mutated cell
line CCRF-CEM. Interestingly, 2 µg/mL of mAb 604.17 preferentially
inhibited the transcriptional activation of the NOTCH1 mutants L1549P,
R1599P, and II1681N as assessed with a validated RPBJ 12xCSL-luciferase
promoter assay. Finally, 15 mg/kg of mAb 604.17 inhibited the tumor
growth of different xenograft cancer models supporting the development
of Notch mAbs as immunotherapeutic tools for different cancers.[35]
Besides Abs directed to NOTCH1 NRR domain, additional probes have been developed to NOTCH2 and NOTCH3.[28]
For example, OMP-59R5 (Tarextumab), was generated by panning the HuCAL
GOLD phage-display library with recombinant NOTCH2 extracellular domain
(EGF1–12) containing the ligand-binding site. OMP-59R5 showed antitumor
activity in breast, ovarian, and small-cell lung cancer.[36]
A phase Ib clinical trial showed that Tarextumab is well tolerated, and
showed a dose-dependent biomarker-driven activity in patients with
small-cell lung cancer (SCLC).[37,38]
The
Blacklow laboratory leveraged the development of inhibitors and
activators NRR mAbs to dissect the dynamics of NOTCH3 activation.[39,40]
Given the prevalence of NOTCH3 activation (ICN3) and the recurrence of
NOTCH3 mutations in different cancer models, including T-ALL, the
authors demonstrated that MOR20350 and MOR20358 inhibited Notch3
signaling in vitro.[41,42] MOR20350 and MOR20358
exhibited an anti-tumor effect using orthotopic xenograft models
representative of cancer carrying a NOTCH3 PEST (MDA-MB468) or NRR
(TALL-1) mutations, respectively.[41]
A second strategy to inhibit Notch signaling is by developing Abs directed against Notch ligands such as DLL1 and DLL4.[43]
For example, OMP-21M18 emerged from murine hybridoma library screen set
to identify DLL4 inhibitors using a Notch-responsive luciferase
reporter assay in HeLa cells.[44] DLL4 has a unique
role in regulating vascular endothelial cell proliferation and
differentiation. Suppression of DLL4-mediated Notch signaling increases
nonproductive angiogenesis but efficiently inhibited tumor growth in
several cancer models.[45] However chronic inhibition
of dll4 showed to alter normal liver endothelial histology in mice,
rats, and cynomolgus monkeys and promotes subcutaneous vascular
neoplasms in rats.[46] Despite safety concerns,
OMP-21M18/Demcizumab entered clinical development, and it has been
investigated in a phase I dose escalation and expansion study in
patients with previously treated solid tumors (RGN-124, NCT01189929).[47]
However, given the lack of clinical responses assessing the role of
OMP-21M18 in combination with paclitaxel plus gemcitabine in treatment-naïve patients with metastatic pancreatic cancer OncoMed
Pharmaceuticals discontinued ongoing demcizumab trials. Similarly to
demcizumab, enoticumab a humanized IgG1 anti-Dll4 was tested in a phase
I trial in ovarian cancers and solid tumors.[48]
Enoticumab was well tolerated (most of the patients experienced
fatigue, headache, hypertension, and nausea) and response to treatment
was confirmed in 2 out 53 patients (5%) treated at 3 mg/kg (one patient
with papillary serous ovarian carcinoma, and one patient with non–small
cell lung cancer) while 16 patients (36%) had a stable disease.48
Demcizumab, enoticumab trials are not extended to patients with
hematological malignancies so far.
]
Targeting the γ-Secretase Complex
Because of its crucial role in Alzheimer's disease pathology, γ-secretase
has been the target of many small molecules that were initially
designed to reduce the generation of Aβ polypeptides in the amyloid
plaques. Among other substrates, the γ-secretase
complex proteolyzes the release of ICN1 and therefore represents a
critical step in the canonical Notch signaling. Thus, inhibitors of
the γ-secretase
complex (GSIs) that target all NOTCH receptors were re-purposed in
cancers where NOTCH1 mutations are common (T-ALL, CLL) and tumor
dependency has been established in preclinical models. For example, in
T-ALL, several studies showed that GSI treatment induces G0/G1 arrest
along with rapid clearance of intracellular NOTCH1.[49-52]
De
Angelo and collaborators completed the first GSI trial in T-ALL in six
adults and two pediatric patients with leukemia (seven with T-ALL)
treated in average for 56 days with MK-0752 a potent inhibitor
developed by Merck & Co. In a T-ALL patient, with an activating
NOTCH1 mutation, the response was transient.[53]
Overall, MK-0752 was poorly tolerated. In fact, most of the patients
suffered from gastrointestinal toxicity, primarily diarrhea, observed
at drug doses of 300 mg/m2.
Subsequent studies showed that the gastrointestinal toxicity was due to
the simultaneous blockade of NOTCH1 and NOTCH2 mediated by GSIs.
Abrogation of the Notch1/2 signaling in the gut leads to severe
intestinal secretory metaplasia, an increase of goblet cells and a
differentiation failure in the crypts of the small intestine[54] suggesting that targeted inhibition of individual receptors might reduce on-target gut toxicity.[28]
The
MK-0752 failure, rushed for the identification of second generations
GSIs with better tolerability profile and of combination strategies to
overcome the limitation showed with the single drug treatment. Real and
colleagues demonstrated that glucocorticoid therapy in combination with
NOTCH1 inhibition by GSIs improved the antileukemic effect of GSIs and
reduced their gut toxicity in vivo.[55,56] GSI
sensitizes steroids resistant T-ALL cell lines and primary patients to
glucocorticoid therapy and induced apoptosis through induction of
BCL2L11. Mice treated with glucocorticoids and a GSI showed decreased
gastrointestinal toxicity compared to animals treated with GSI alone.
Steroids mediate the induction of cyclin D2 (CCND2), a cyclin
associated with cell cycle progression, and by the down-regulation of
Kruppel-Like Factor 4 (KLF4), a negative regulator of the cell cycle
that is required for goblet cell differentiation.[14,55]
In addition, Cullion and collaborators demonstrated that intermittent
GSI dosing with drug holiday largely avoided gastrointestinal toxicity
while maintaining efficacy in a mouse T-ALL model.[57,58]
However,
gut toxicity is not the only off-target effect seen in GSI treated
patients, raising additional concerns on chronic inhibition of
wild-type NOTCH1. In two early-terminated phase III trials, LY450139
(semagacestat), failed to achieve the primary endpoints (improvement in
the cognition and the ability to complete activities of daily living)
in patients with mild-to-moderate Alzheimer's disease.[59]
Data showed that semagacestat was associated with an increased risk of
skin cancer compared with those who received placebo, likely due to
inhibition of Notch in the skin by chronic GSI administration
consistent with the tumor-suppressor role of Notch signaling in this
tissue.[60,61] In addition, recent studies suggested
that Notch signaling blockade might increase the risk of developing
lung squamous cell carcinoma (SCC).[62] Whether this
risk will be ameliorated by intermittent, pulsed therapy with GSI, as
would be the schedule in cancer-directed therapy, is still to be
determined.[14]
An additional GSI that reached
clinical development is PF-03084014/Nirogacestat a noncompetitive,
reversible GSI developed by Pfizer.[63] PF-03084014
induced an anti-leukemic effect in vitro and in vivo in T-ALL cell
lines expressing mutant NOTCH1. An intermittent dosing schedule of
PF-03084014 and the addition of glucocorticoids attenuated
Notch-dependent gastrointestinal toxicity by reducing the loss of body
weight in an HBP-ALL T-ALL xenograft model[63]
confirming previous Cullion’s observations. PF-03084014 induces
selective apoptosis in primary CLL cells carrying NOTCH1 mutations and
synergize with fludarabine in a stroma coculture model system.[64]
In a phase I trial aimed to determine the safety profile and maximum
tolerated dose (MTD) of PF-03084014, one out of eight
relapsed/refractory T-ALL patients achieved a complete remission.[65]
Knoechel
and colleagues reported a complete hematological response in a patient
with early T-cell precursor acute lymphoblastic leukemia (ETP-ALL)
carrying a NOTCH1 mutation treated with the GSI developed by the
Bristol-Myers-Squibb 906024. A phase I trial, CA216002, confirmed this
encouraging result and demonstrated the safety of BMS-906024
administered on weekly dosing (4-6 mg) in 25 pediatric patients with
T-ALL or T-cell lymphoblastic lymphoma.[66] This study was the first
trial reporting multiple responses to GSI inhibition, including a
complete response and one partial response. Overall, 32% of the
patients showed at least a 50% reduction in bone marrow (BM) blasts
with tolerable side effects.[66] Interestingly, in
pre-clinical studies, BMS-906024 enhanced the anti-leukemic activity of
Ibrutinib in B-CLL cells in vitro by inhibiting ICN1 activation and
consequently the transcription of its targets such as c-MYC.[67]
An alternative strategy to modulate γ-secretase activity is by developing mAbs directed to functional components of this complex. The γ-secretase
complex comprises a catalytic core formed by presenilin 1 and
presenilin 2 (PS1 and PS2) and three accessory proteins: anterior
pharynx-defective 1 (APH-1), nicastrin (NCT), and presenilin enhancer
protein 2 (PEN2).[68] For example, Hayashi and
colleagues reported the identification of two mAbs A5226A and A5201A
directed against the extracellular domain of NCT.[69,70] A5226A inhibited γ-secretase
activity by competing with the NCT substrate binding in vitro. In
addition, A5226A inhibited the proliferation of a NOTCH1 mutated T-ALL
cell line, DND41, and prevented ICN1 cleavage. In a xenograft model of
DND41, A5226A administered at 50 mg/Kg/day reduced cancer cells growth
in vivo.[69]
As discussed above, several GSIs showed preclinical activity and have entered late development,[71] limitations include lack of substrate selectivity and toxicities.[72]
In addition, genetic and epigenetic mechanisms of resistance partially
explained the lack of successful clinical translation on a large scale.
To identify mechanisms of resistance to NOTCH1 inhibition in T-ALL, the
laboratory of Dr. Ferrando analyzed the global gene expression
signatures associated with a sensitivity of resistance to GSI. They
demonstrated that the transcriptional suppression of PTEN was
associated with resistance to GSI treatment in T-ALL cell lines.
Protein analysis and mutation sequencing showed the absence, or the
marked reduction of PTEN at the protein level and biallelic PTEN
mutation in resistant T-ALL cell lines.[73,74]
Knoechel
and collaborators described an additional mechanism of tolerance to GSI
therapy. In this work, the authors identified from in vitro long-term
culture under GSI positive selections a subpopulation of GSI-tolerant
T-ALL cells called “persister”. They described that resistance to GSI
was reversible after the drug’s withdrawal; thus, they speculated the
existence of an epigenetic mechanism of drug resistance. Therefore,
they performed a short hairpin RNA (shRNA) screen targeting genes
involved in chromatin regulation. Among top hits, which preferentially
impaired the viability of “persister” cells while sparing the naïve
population, they identified the BET (bromodomain and extra-terminal
domain) family, BRD4. Consistently “persister” cells were more
sensitive to BRD4 inhibition (JQ1) in vitro and combination therapy
targeting “naïve” (GSI) and “persister” (JQ1) was significantly more
effective in T-ALL xenotransplant models in vivo.[75]
Targeting NOTCH Trafficking
As
we described above, NOTCH1 is a rational therapeutic target in several
hematological malignancies, but as a mutated transcription factor, it
poses a drug discovery challenge. Several groups contributed to the
development of a program to overcome limitations associated with the
targeting of transcription factors (e.g. NOTCH1)[76-81] or resistance to target therapy.[82-84] For example, we completed a gene expression-based high-throughput small molecule (GE-HTS)[49,85] and a cDNA overexpression screen using cell-based assays reporting Notch transcriptional activity.[86]
To enrich for targets that preferentially impair NOTCH1 receptor
bearing HD mutations (NRR), we deliberately selected to screen against
a human T-ALL cell line (DND41), which carries a clinically relevant
activating mutation in the HD of NOTCH1 along with a PEST domain
deletion (L1594PΔPEST) and secondly to identify gene products that
would enhance the activation of a transcriptional reporter downstream
of a mutant NOTCH1 receptor frequently identified in T-ALL patients
(L1601PΔPEST). Several ion flux modulators or genes encoding for ion
channels or pumps scored as hits in the small molecules or the cDNA
screens, respectively. One of the top compound hits was thapsigargicin,
an analog of thapsigargin, which is a non competitive inhibitor of the
sarco/endoplasmic reticulum Ca2+-ATPase
(SERCA). Among the top cDNA hits were ATP2A1, ATP2A2, and ATP2A3, which
encode SERCA1, SERCA2, and SERCA3, respectively. We next showed that
SERCA inhibition impairs the trafficking of mutated NOTCH1 receptors
and induces a G0/G1 arrest in NOTCH1-mutated human T-ALL cells (Figure 1B). Thapsigargin had on-target activity in mouse models of human T-ALL and also interfered with Notch signaling in Drosophila.[76,87] Remarkably, thapsigargin preferentially inhibited mutated NOTCH1 receptors.[76]
This selectivity provides a therapeutic window not observed before with
GSIs or most antibody-based approaches that are equipotent inhibitors
of mutated and wild type (WT) receptors. Subsequent independent studies
confirmed our original observation and demonstrated that thapsigargin
alone or in combination with mAb 604.107 inhibited “gain of function”
mutants associated with T-ALL such as L1594P, R1599P and I168N.[35]
Thapsigargin is an organic heterotricyclic compound that is a hexa-γoxygenated 6,7-guaianolide isolated from the roots of Thapsia garganica. Thapsigargin inhibits SERCA-mediated calcium (Ca2+) uptake leading to a depletion of the endoplasmic reticulum (ER) Ca2+ storage and sustained elevation of cytosolic Ca2+
triggering ER stress, [76] unfolded protein response (UPR), and different
cellular pathways that can cause cell death. This
general mechanism of cytotoxicity to develop SERCA inhibitors for
cancer therapies has been leveraged . For example, SERCA has been identified as an emerging
target in the treatment of prostate cancer.[88] SERCA channels are critical to maintaining intracellular Ca2+
homeostasis in all cell types. Thus, the direct delivery of
thapsigargin to animals or humans might be expected to incur cardiac
toxicity secondary to Ca2+
ion shifts. A strategy to prevent a systemic cytotoxic effect by
inhibiting SERCA is by creating inactive pro-drugs that are activated
in a histo-specific manner.[89] This, for example, is the mode of action of mipsagargin,[90,91] a TG soluble prodrug undergoing clinical trials for solid tumor.[89]
In
the past, we imagined a general strategy for efficient TG delivery
leveraging the dependency to folate metabolism of leukemia cells and
developed a folate-TG derivative compound to transfer the inhibitor
specifically to the T-ALL cells.[92] We showed that
the 8-O-debutanoylthapsigargin, a cytotoxic TG analog, retained the
anti-leukemia specificity toward mutant NOTCH1 in T-ALL cell lines.
Thus, we linked the carboxylate of folic acid to the C8-alcohol of 8-O-debutanoylthapsigargin, to generate the folate-thapsigargin conjugate
named JQ-FT. We demonstrated that JQ-FT inhibits NOTCH1 in vitro in
multiple T-ALL models and in vivo on a syngeneic T-ALL mouse model
carrying a NOTCH1 L1601P ΔPEST a common mutation observed in the human
disease.[92] In the Notch arena, JQ-FT is the
first-in-class NOTCH1 inhibitor with dual selectivity: leukemia over
normal and NOTCH1-mutant over wild type receptors.
In the recent
past, several putative SERCA inhibitors have been described. However,
only a few have been tested in Notch-dependent diseases. Ford and
colleagues demonstrated that the natural tricyclic clerodane diterpene
casearin J (CJ),[93] can affect the Notch1 pathway in
human T-ALL cells. CJ reduced cell surface expression of NOTCH1
receptors, prevented the formation of the cleaved ICN1 molecules, which
resulted in the transcriptional inhibition of Notch targets such as
MYC, HES1. The authors showed that CJ inhibits SERCA protein causing a
rise of intracellular Ca2+ and depletion of the ER Ca2+
storage. This ion shift concentration increases reactive oxygen species
(ROS) and ultimately leads to apoptosis in T-ALL cells.[93]
However, while the authors claimed selectivity toward HD-mutations,
they did not demonstrate the lack of CJ activity in a large panel of
wild type T-ALL models. In addition, is not clear whether CJ causes an
accumulation of full-length NOTCH1, as for other SERCA inhibitors,[76] suggesting that different interactions in the SERCA binding site may be responsible for the effect on Ca2+ and consequently on Notch activation.
Ethyl
2-Amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate
(CXL017) is a recently synthesized SERCA inhibitor tested in multiple
leukemia cell lines that acquired multidrug resistance through
different mechanisms, including T-ALL.[94] Additional
studies demonstrated that CXL017 synergizes with other SERCA inhibitors
including thapsigargin and cyclopiazonic acid indicating that CXL017
may bind SERCA at a unique allosteric site[95] pointing to the potential of developing new classes of SERCA modulators.
In our original GE-HTS screen, multiple compounds reported modulating Ca2+ ion flux scored as dose-dependent Notch pathway inhibitors including ionomycin, salinomycin, and bepridil.[76] Thus we initially extended testing the FDA approved Ca2+ antagonist bepridil in T-ALL.[96]
In vitro, bepridil reduced ICN1 and consequently caused a phenotype
consistent with Notch abrogation in this tumor. While we can reach this
effect at the plasma level concentration achievable in human, we did
not demonstrate an effect in T-ALL orthographs, and we halted further
experiments.[96] However, because we showed a
transcriptional overlap between the NOTCH1 “Off” signatures in T-ALL
and CLL, we re-purposed bepridil for B-cell malignancies.[97] In CLL bepridil exerted an anti-leukemia activity in vivo associated with NOTCH1 inhibition.[97]
Similar to thapsigargin, histological analysis of the gut showed normal
goblet cell number with preservation of the architecture and
proliferation of the intestinal epithelium suggesting a lack of
combined NOTCH1 and NOTCH2 inhibition in this tissue. This result
suggests Ca2+ mediated inhibition of Notch signaling may overcome the limitation associated with γ-secretase inhibition.
An
additional strategy to alter NOTCH trafficking is by modulating the
protein O-fucosyltransferase-1. POFUT1 catalyzes the addition of
O-linked fucose to the EGF-repeat domains of the NOTCH receptor that is
required for NOTCH activation.[98] McMillan and
colleagues showed that CRISPR/Cas9 mediated POFUT1 knockout in U2OS
cells suppresses Notch activation signaling associated with type I and
II mutations.[99] Interestingly, NOTCH1 protein does
not mature in the CRISPR-engineered U2OS cells lacking POFUT1, a
phenotype that mimics closely TG inhibition.
Targeting NOTCH Degradation
NOTCH
is a short-lived protein and undergoes degradation mainly through an
E3-ligase (Fbw7) ubiquitin-mediated pathway controlled by the PEST
domain. As we described above, disruption of the PEST domain leads to
an increase in ICN half-life.[25] In recent work,
Koyama and colleagues demonstrated that the proteasome inhibitor,
bortezomib, repressed the transcription of NOTCH1 and of its downstream
targets including HES1, GATA3, RUNX3 and CYLD in MOLT4, JURKAT and CEM
T-ALL cell lines.[100]
Drug combination studies
revealed that bortezomib showed synergistic or additive effects with
key drugs to treat T-ALL such as dexamethasone, doxorubicin, and
cyclophosphamide. The synergistic effect of bortezomib and
dexamethasone was confirmed at NOTCH1 protein expression level and
later in vivo using a murine MOLT-4 T-ALL cell xenograft model.[100]
This study supported the rationale of an ongoing clinical trial
assessing the role of bortezomib in combination with different
chemotherapy regimen (NCT02112916) in younger patients with newly
diagnosed T-ALL or stage II-IV T-cell lymphoblastic lymphoma.
In
parallel, Bertaina and colleagues tested bortezomib in combination with
chemotherapy in 30 and 7 children with B-cell precursor (BCP) and
T-cell ALL, respectively.[101] Bortezomib (1.3 mg/m2/dose)
was administered intravenously twice a week x 2 with a chemotherapy
regimen containing dexamethasone, doxorubicin, vincristine, and
pegylated asparaginase. Twenty-two of 30 BCP-ALL patients (73,3%) and
5/7 patients (71%) with T-cell ALL achieved CR/CRp. The 2-year overall
survival (OS) was 31,3% while patients that achieved an MRD response
had a 2-year OS of 68·4%.[101] These data suggest that bortezomib may represent a clinically effective option in NOTCH1 mutated T-ALL patients.
In CLL, Notch2 signaling appears to have a constitutive role in promoting cell survival and CD23 expression.[102,103] Several studies showed that B-CLL undergoes apoptosis upon proteasome inhibitors treatment.[104,105]
However, Duecheler and colleagues demonstrated that bortezomib and
MG132 efficiently induced apoptosis in B-CLLs in vitro by inhibiting
NOTCH2 transactivation and repressing CD23 expression.[106] Similarly, in MCL, several studies demonstrated the effects of proteasome inhibition on several intracellular mechanisms.[107] For example, bortezomib showed to induce cell cycle arrest and apoptosis by inhibition of NF-kB,[108] inhibition of the protein kinase CK2,[109,110] the depolarization of the mitochondria membrane, ROS release, and the production of pro-apoptotic proteins (NOXA).[111] In addition, several pre-clinical studies demonstrated the synergist activity of bortezomib with other antineoplastic agents[112,113] including the HDAC inhibitor vorinostat (SAHA),[114] idelalisib,[115] and the anti-CD20 mAb rituximab.[116]
While many clinical trials confirmed that combining bortezomib with
other anti-lymphoma therapies is feasible effective none at the moment
focused on the role of Notch signaling mediating the efficacy or
resistant to therapy.
Targeting ICN1 Complex
As
described above, activation of NOTCH1 receptor results in a sequence of
cleavages that cause the release of ICN1. Following translocation to
the nucleus, ICN1 forms a ternary complex with the transcriptional
repressor CSL (CBF-1, Suppressor of Hairless and Lag-1) co-activators
of the Mastermind-like family (MAML1-3 in humans) bound to DNA. Thus,
Moellering and colleagues developed a cellular penetrant, soluble
α-Helix-constrained “stapled” peptide derived from mastermind-like 1,
SAHM1 that can bind the ICN-CSL complex. Similarly to GSI, SAHM1
produced a transcriptional signature of NOTCH gene repression in human
and murine T-ALL cells. Direct blockade of NOTCH-CSL transcriptional
complex reduced NOTCH-specific anti- proliferative effects in human
T-ALL cell lines and in a bioluminescent murine model of T-ALL.[117]
While
this approach holds the premises to be more specific for Notch compared
to GSIs, which also affect the cleavage of different cellular
substrates, its clinical translation is hampered by the lack of
pharmacokinetics and pharmacodynamics studies.
Recently Cellestia
Biotech AG developed CB-103 a small molecule protein-protein
interaction (PPI) inhibitor able to target assembly of the Notch
transcription complex in the cell nucleus leading to down-regulation of
Notch target genes (c-MYC, CCND1, HES1) and inhibition of Notch
signaling independently of Notch mechanisms of activation. This
pan-Notch inhibitor has shown preclinical activity in a variety of
solid tumors and leukemia models. In preclinical studies CB-103
inhibited the proliferation of various cancer cell lines including
T-ALL with known NOTCH1 mutational status (RPMI-8402 and KOPT-K1)
compared to the GSI 4929097. Both ICN1, transmembrane NOTCH1 and
full-length decrease upon CB-103 treatment consistent with a mechanism
of transcriptional inhibition.[118] Spriano and
colleagues extended testing CB-103 in a collection of 61 B and T cell
lymphoma cell lines. CB-103 presented a median IC50 above 20 µM across
the whole panel of lymphoma cell lines (range from 400 nM to > 20
µM), without significant differences among lymphoma subtypes.[119]
Sensitive lines (IC50 < 10 µM) presented a gene expression signature
significantly enriched with genes involved in the
epithelial-mesenchymal transition, a Notch-related process.[119]
A multicenter open-label, non randomised phase I-II clinical trial
(CB-103-C-101) is ongoing, enrolling patients with advanced, refractory
or metastatic solid tumors and hematological malignancies for whom no
standard therapy exists.[120] Notch mutational
status or expression is not key inclusion criteria of the study but it
stands among the exploratory analysis suggesting that, as in other
previous studies, responses in Notch mutated cases may be few.
Conclusions
In
the last two decades, we have seen significant improvements in T-ALL,
CLL and MCL survival. However, a significant number of patients relapse
or rapidly became resistant to available therapeutic options. Thus, the
development of a Notch targeted approach appears a rational strategy to
modulate a pathway on which these cancer cells rely on to survive.
Despite γ-secretase
inhibitors experienced several roadblocks in their development we are
achieving a better characterization of disease's pathways that will
facilitate the development of mutant selective of context-dependent
inhibitors for these aggressive tumors. Furthermore, the development of
Notch isoform selective small molecules along with re-defined
therapeutic schedule will overcome the hurdle associated with the
off-target toxicities seen with the chronic inhibition of wild type
NOTCH1 and NOTCH2.
Acknowledgments
This
work was supported by an AIRC Start-up Investigator Grant (n. 17107
G.R.), Fondazione Cariparma (3576/2017, 0180/2018 G.R.), Fondazione
Grande Ale Onlus (G.R.), Fondazione Umberto Veronesi Post-doctoral
Fellowship (C.S.).
References
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT,
Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G.
Discovery and saturation analysis of cancer genes across 21 tumour
types. Nature. 2014;505(7484):495-501. https://doi.org/10.1038/nature12912 PMid:24390350 PMCid:PMC4048962
- Artavanis-Tsakonas
S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal
integration in development. Science. 1999;284(5415):770-776. https://doi.org/10.1126/science.284.5415.770 PMid:10221902
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216-233. https://doi.org/10.1016/j.cell.2009.03.045 PMid:19379690 PMCid:PMC2827930
- Gordon
WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, Blacklow
SC. Structure of the Notch1-negative regulatory region: implications
for normal activation and pathogenic signaling in T-ALL. Blood.
2009;113(18):4381-4390. https://doi.org/10.1182/blood-2008-08-174748 PMid:19075186 PMCid:PMC2676092
- Gordon
WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC.
Structural basis for autoinhibition of Notch. Nat Struct Mol Biol.
2007;14(4):295-300. https://doi.org/10.1038/nsmb1227 PMid:17401372
- Brou
C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P,
Black RA, Israel A. A novel proteolytic cleavage involved in Notch
signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell.
2000;5(2):207-216. https://doi.org/10.1016/S1097-2765(00)80417-7
- Moretti J, Brou C. Ubiquitinations in the notch signaling pathway. Int J Mol Sci. 2013;14(3):6359-6381. https://doi.org/10.3390/ijms14036359 PMid:23519106 PMCid:PMC3634445
- Sanchez-Irizarry
C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit
heterodimerization and prevention of ligand-independent proteolytic
activation depend, respectively, on a novel domain and the LNR repeats.
Mol Cell Biol. 2004;24(21):9265-9273. https://doi.org/10.1128/MCB.24.21.9265-9273.2004 PMid:15485896 PMCid:PMC522238
- De
Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS,
Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A
presenilin-1-dependent gamma- secretase-like protease mediates release
of Notch intracellular domain. Nature. 1999;398(6727):518-522. https://doi.org/10.1038/19083 PMid:10206645
- Ellisen
LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J. TAN-1,
the human homolog of the Drosophila notch gene, is broken by
chromosomal translocations in T lymphoblastic neoplasms. Cell.
1991;66(4):649-661. https://doi.org/10.1016/0092-8674(91)90111-B
- Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program. 2009:353-361. https://doi.org/10.1182/asheducation-2009.1.353 PMid:20008221 PMCid:PMC2847371
- Weng
AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C,
Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human
T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271. https://doi.org/10.1126/science.1102160 PMid:15472075
- Malecki
MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC,
Blacklow SC. Leukemia-associated mutations within the NOTCH1
heterodimerization domain fall into at least two distinct mechanistic
classes. Mol Cell Biol. 2006;26(12):4642-4651. https://doi.org/10.1128/MCB.01655-05 PMid:16738328 PMCid:PMC1489116
- Roti G, Stegmaier K. Targeting NOTCH1 in hematopoietic malignancy. Crit Rev Oncog. 2011;16(1-2):103-115. https://doi.org/10.1615/CritRevOncog.v16.i1-2.100
- Di
Ianni M, Baldoni S, Rosati E, Ciurnelli R, Cavalli L, Martelli MF,
Marconi P, Screpanti I, Falzetti F. A new genetic lesion in B-CLL: a
NOTCH1 PEST domain mutation. Br J Haematol. 2009;146(6):689-691. https://doi.org/10.1111/j.1365-2141.2009.07816.x PMid:19604236
- Puente
XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G,
Jares P, Bea S, Gonzalez-Diaz M, Bassaganyas L, Baumann T, Juan M,
Lopez-Guerra M, Colomer D, Tubio JM, Lopez C, Navarro A, Tornador C,
Aymerich M, Rozman M, Hernandez JM, Puente DA, Freije JM, Velasco G,
Gutierrez-Fernandez A, Costa D, Carrio A, Guijarro S, Enjuanes A,
Hernandez L, Yague J, Nicolas P, Romeo-Casabona CM, Himmelbauer H,
Castillo E, Dohm JC, de Sanjose S, Piris MA, de Alava E, San Miguel J,
Royo R, Gelpi JL, Torrents D, Orozco M, Pisano DG, Valencia A, Guigo R,
Bayes M, Heath S, Gut M, Klatt P, Marshall J, Raine K, Stebbings LA,
Futreal PA, Stratton MR, Campbell PJ, Gut I, Lopez-Guillermo A,
Estivill X, Montserrat E, Lopez-Otin C, Campo E. Whole-genome
sequencing identifies recurrent mutations in chronic lymphocytic
leukaemia. Nature. 2011;475(7354):101-105. https://doi.org/10.1038/nature10113 PMid:21642962 PMCid:PMC3322590
- Arruga
F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, Laurenti L,
D'Arena G, Jaksic O, Inghirami G, Rossi D, Gaidano G, Deaglio S.
Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia.
Leukemia. 2014;28(5):1060-1070. https://doi.org/10.1038/leu.2013.319 PMid:24170027
- Kridel
R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J,
Jenkins C, Cochrane C, Ben-Neriah S, Tan K, Morin RD, Opat S, Sehn LH,
Connors JM, Marra MA, Weng AP, Steidl C, Gascoyne RD. Whole
transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle
cell lymphoma. Blood. 2012;119(9):1963-1971. https://doi.org/10.1182/blood-2011-11-391474 PMid:22210878
- Bea
S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P,
Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di
Croce L, Puente DA, Lopez- Guerra M, Moros A, Roue G, Aymerich M,
Villamor N, Colomo L, Martinez A, Valera A, Martin-Subero JI, Amador V,
Hernandez L, Rozman M, Enjuanes A, Forcada P, Muntanola A, Hartmann EM,
Calasanz MJ, Rosenwald A, Ott G, Hernandez-Rivas JM, Klapper W, Siebert
R, Wiestner A, Wilson WH, Colomer D, Lopez-Guillermo A, Lopez- Otin C,
Puente XS, Campo E. Landscape of somatic mutations and clonal evolution
in mantle cell lymphoma. Proc Natl Acad Sci U S A.
2013;110(45):18250-18255. https://doi.org/10.1073/pnas.1314608110 PMid:24145436 PMCid:PMC3831489
- Fabbri
G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, Yan XJ,
Kieso Y, Rossi D, Gaidano G, Chiorazzi N, Ferrando AA, Dalla-Favera R.
Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia.
Proc Natl Acad Sci U S A. 2017;114(14):E2911-E2919. https://doi.org/10.1073/pnas.1702564114 PMid:28314854 PMCid:PMC5389283
- Baliakas
P, Hadzidimitriou A, Sutton LA, Rossi D, Minga E, Villamor N, Larrayoz
M, Kminkova J, Agathangelidis A, Davis Z, Tausch E, Stalika E,
Kantorova B, Mansouri L, Scarfo L, Cortese D, Navrkalova V,
Rose-Zerilli MJ, Smedby KE, Juliusson G, Anagnostopoulos A, Makris AM,
Navarro A, Delgado J, Oscier D, Belessi C, Stilgenbauer S, Ghia P,
Pospisilova S, Gaidano G, Campo E, Strefford JC, Stamatopoulos K,
Rosenquist R, European Research Initiative on CLL. Recurrent mutations
refine prognosis in chronic lymphocytic leukemia. Leukemia.
2015;29(2):329-336. https://doi.org/10.1038/leu.2014.196 PMid:24943832
- Inamdar
AA, Goy A, Ayoub NM, Attia C, Oton L, Taruvai V, Costales M, Lin YT,
Pecora A, Suh KS. Mantle cell lymphoma in the era of precision
medicine-diagnosis, biomarkers and therapeutic agents. Oncotarget.
2016;7(30):48692-48731. https://doi.org/10.18632/oncotarget.8961 PMid:27119356 PMCid:PMC5217048
- Rossi
D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, Marasca R,
Laurenti L, Bruscaggin A, Cerri M, Monti S, Cresta S, Fama R, De Paoli
L, Bulian P, Gattei V, Guarini A, Deaglio S, Capello D, Rabadan R,
Pasqualucci L, Dalla-Favera R, Foa R, Gaidano G. Mutations of NOTCH1
are an independent predictor of survival in chronic lymphocytic
leukemia. Blood. 2012;119(2):521-529. https://doi.org/10.1182/blood-2011-09-379966 PMid:22077063 PMCid:PMC3257017
- Schmitz
R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S,
Kasbekar M, Young RM, Shaffer AL, Hodson DJ, Xiao W, Yu X, Yang Y, Zhao
H, Xu W, Liu X, Zhou B, Du W, Chan WC, Jaffe ES, Gascoyne RD, Connors
JM, Campo E, Lopez-Guillermo A, Rosenwald A, Ott G, Delabie J, Rimsza
LM, Tay Kuang Wei K, Zelenetz AD, Leonard JP, Bartlett NL, Tran B,
Shetty J, Zhao Y, Soppet DR, Pittaluga S, Wilson WH, Staudt LM.
Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J
Med. 2018;378(15):1396-1407. https://doi.org/10.1056/NEJMoa1801445 PMid:29641966 PMCid:PMC6010183
- Chiang
MY, Radojcic V, Maillard I. Oncogenic Notch signaling in T-cell and
B-cell lymphoproliferative disorders. Curr Opin Hematol.
2016;23(4):362-370. https://doi.org/10.1097/MOH.0000000000000254 PMid:27135981 PMCid:PMC4962559
- Gu
Y, Masiero M, Banham AH. Notch signaling: its roles and therapeutic
potential in hematological malignancies. Oncotarget.
2016;7(20):29804-29823. https://doi.org/10.18632/oncotarget.7772 PMid:26934331 PMCid:PMC5045435
- Cuesta-Mateos
C, Alcaraz-Serna A, Somovilla-Crespo B, Munoz-Calleja C. Monoclonal
Antibody Therapies for Hematological Malignancies: Not Just
Lineage-Specific Targets. Front Immunol. 2017;8:1936. https://doi.org/10.3389/fimmu.2017.01936 PMid:29387053 PMCid:PMC5776327
- Wu
Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D,
Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki
S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan
CA, Hymowitz SG, Siebel CW. Therapeutic antibody targeting of
individual Notch receptors. Nature. 2010;464(7291):1052-1057. https://doi.org/10.1038/nature08878 PMid:20393564
- Yuan
X, Wu H, Xu H, Xiong H, Chu Q, Yu S, Wu GS, Wu K. Notch signaling: an
emerging therapeutic target for cancer treatment. Cancer Lett.
2015;369(1):20-27. https://doi.org/10.1016/j.canlet.2015.07.048 PMid:26341688
- Bellavia
D, Palermo R, Felli MP, Screpanti I, Checquolo S. Notch signaling as a
therapeutic target for acute lymphoblastic leukemia. Expert Opin Ther
Targets. 2018;22(4):331-342. https://doi.org/10.1080/14728222.2018.1451840 PMid:29527929
- Aste-Amezaga
M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, Huang L, Vitelli
S, Vo KT, Haytko P, Zhao JZ, Baleydier F, L'Heureux S, Wang H, Gordon
WR, Thoryk E, Andrawes MB, Tiyanont K, Stegmaier K, Roti G, Ross KN,
Franlin LL, Wang H, Wang F, Chastain M, Bett AJ, Audoly LP, Aster JC,
Blacklow SC, Huber HE. Characterization of Notch1 antibodies that
inhibit signaling of both normal and mutated Notch1 receptors. PLoS
One. 2010;5(2):e9094. https://doi.org/10.1371/journal.pone.0009094 PMid:20161710 PMCid:PMC2817004
- Agnusdei
V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, Gurney A, Hoey
T, Seganfreddo E, Basso G, Valtorta S, Moresco RM, Amadori A,
Indraccolo S. Therapeutic antibody targeting of Notch1 in T-acute
lymphoblastic leukemia xenografts. Leukemia. 2014;28(2):278-288. https://doi.org/10.1038/leu.2013.183 PMid:23774673
- Höring
E, Colom Sanmarti B, Xargay-Torrent S, Aulitzky WE, van der Kuip H,
Campo E, López-Guerra M, Colomer D. Notch1 Signaling in
<em>NOTCH1</em>-Mutated Mantle Cell Lymphoma Depends on
DLL4 and Is a Potential Target for Specific Antibody Therapy. Blood.
2016;128:1846-1846.
- Casulo
C, Ruan J, Dang NH, Gore L, Diefenbach C, Beaven AW, Castro JE, Porcu
P, Faoro L, Dupont J, Kapoun A, Wang M, McGuire K, Flinn IW. Safety and
Preliminary Efficacy Results of a Phase I First-in-Human Study of the
Novel Notch-1 Targeting Antibody Brontictuzumab (OMP-52M51)
Administered Intravenously to Patients with Hematologic Malignancies.
Blood. 2016;128:5108-5108.
- Sharma
A, Gadkari RA, Ramakanth SV, Padmanabhan K, Madhumathi DS, Devi L,
Appaji L, Aster JC, Rangarajan A, Dighe RR. A novel Monoclonal Antibody
against Notch1 Targets Leukemia-associated Mutant Notch1 and Depletes
Therapy Resistant Cancer Stem Cells in Solid Tumors. Sci Rep.
2015;5:11012. https://doi.org/10.1038/srep11012 PMid:26046801 PMCid:PMC4457015
- Yen
WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, Henner WR,
Meisner R, Sato A, Shah J, Tang T, Wallace B, Wang M, Zhang C, Kapoun
AM, Lewicki J, Gurney A, Hoey T. Targeting Notch signaling with a
Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and
decreases tumor-initiating cell frequency. Clin Cancer Res.
2015;21(9):2084-2095. https://doi.org/10.1158/1078-0432.CCR-14-2808 PMid:25934888
- MC
P. Final results of phase Ib of tarextumab (TRXT, OMP-59R5,
anti-Notch2/3) in combination with etoposide and platinum (EP) in
patients (pts) with untreated extensive- stage small-cell lung cancer
(ED-SCLC). J Clin Oncol. 2015;33:abstr 7508. https://doi.org/10.1200/jco.2015.33.15_suppl.7508
- Smith
DC, Chugh R, Patnaik A, Papadopoulos KP, Wang M, Kapoun AM, Xu L,
Dupont J, Stagg RJ, Tolcher A. A phase 1 dose escalation and expansion
study of Tarextumab (OMP-59R5) in patients with solid tumors. Invest
New Drugs. 2018. https://doi.org/10.1007/s10637-018-0714-6 PMCid:PMC6440937
- Tiyanont
K, Wales TE, Siebel CW, Engen JR, Blacklow SC. Insights into Notch3
activation and inhibition mediated by antibodies directed against its
negative regulatory region. J Mol Biol. 2013;425(17):3192-3204. https://doi.org/10.1016/j.jmb.2013.05.025 PMid:23747483 PMCid:PMC3751422
- Xu
X, Choi SH, Hu T, Tiyanont K, Habets R, Groot AJ, Vooijs M, Aster JC,
Chopra R, Fryer C, Blacklow SC. Insights into Autoregulation of Notch3
from Structural and Functional Studies of Its Negative Regulatory
Region. Structure. 2015;23(7):1227-1235. https://doi.org/10.1016/j.str.2015.05.001 PMid:26051713 PMCid:PMC4497832
- Bernasconi-Elias
P, Hu T, Jenkins D, Firestone B, Gans S, Kurth E, Capodieci P,
Deplazes- Lauber J, Petropoulos K, Thiel P, Ponsel D, Hee Choi S,
LeMotte P, London A, Goetcshkes M, Nolin E, Jones MD, Slocum K, Kluk
MJ, Weinstock DM, Christodoulou A, Weinberg O, Jaehrling J, Ettenberg
SA, Buckler A, Blacklow SC, Aster JC, Fryer CJ. Characterization of
activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia
and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene.
2016;35(47):6077-6086. https://doi.org/10.1038/onc.2016.133 PMid:27157619 PMCid:PMC5102827
- Choi
SH, Severson E, Pear WS, Liu XS, Aster JC, Blacklow SC. The common
oncogenomic program of NOTCH1 and NOTCH3 signaling in T-cell acute
lymphoblastic leukemia. PLoS One. 2017;12(10):e0185762. https://doi.org/10.1371/journal.pone.0185762 PMid:29023469 PMCid:PMC5638296
- Briot
A, Iruela-Arispe ML. Blockade of specific NOTCH ligands: a new
promising approach in cancer therapy. Cancer Discov. 2015;5(2):112-114.
https://doi.org/10.1158/2159-8290.CD-14-1501 PMid:25656896 PMCid:PMC4342039
- Hoey
T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M,
Lazetic S, Park IK, Sato A, Satyal S, Wang X, Clarke MF, Lewicki J,
Gurney A. DLL4 blockade inhibits tumor growth and reduces
tumor-initiating cell frequency. Cell stem cell. 2009;5(2):168- 177. https://doi.org/10.1016/j.stem.2009.05.019 PMid:19664991
- Ridgway
J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts
RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo JA, de Sauvage
F, Plowman G, Yan M. Inhibition of Dll4 signalling inhibits tumour
growth by deregulating angiogenesis. Nature. 2006;444(7122):1083-1087. https://doi.org/10.1038/nature05313 PMid:17183323
- Yan
M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, Niessen K,
Plowman GD. Chronic DLL4 blockade induces vascular neoplasms. Nature.
2010;463(7282):E6-7. https://doi.org/10.1038/nature08751 PMid:20147986
- Smith
DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, Kapoun AM,
Xu L, Dupont J, Sikic B. A phase I dose escalation and expansion study
of the anticancer stem cell agent demcizumab (anti-DLL4) in patients
with previously treated solid tumors. Clin Cancer Res.
2014;20(24):6295-6303. https://doi.org/10.1158/1078-0432.CCR-14-1373 PMid:25324140
- Chiorean
EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA,
Adriaens L, Liu L, Kao RJ, DiCioccio AT, Kostic A, Leek R, Harris A,
Jimeno A. A Phase I First-in-Human Study of Enoticumab (REGN421), a
Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients
with Advanced Solid Tumors. Clin Cancer Res. 2015;21(12):2695-2703. https://doi.org/10.1158/1078-0432.CCR-14-2797 PMid:25724527
- Weng
AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C,
Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human
T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271. https://doi.org/10.1126/science.1102160 PMid:15472075
- De
Keersmaecker K, Lahortiga I, Mentens N, Folens C, Van Neste L, Bekaert
S, Vandenberghe P, Odero MD, Marynen P, Cools J. In vitro validation of
gamma-secretase inhibitors alone or in combination with other
anti-cancer drugs for the treatment of T-cell acute lymphoblastic
leukemia. Haematologica. 2008;93(4):533-542. https://doi.org/10.3324/haematol.11894 PMid:18322257
- Palomero
T, Barnes KC, Real PJ, Glade Bender JL, Sulis ML, Murty VV, Colovai AI,
Balbin M, Ferrando AA. CUTLL1, a novel human T-cell lymphoma cell line
with t(7;9) rearrangement, aberrant NOTCH1 activation and high
sensitivity to gamma-secretase inhibitors. Leukemia.
2006;20(7):1279-1287. https://doi.org/10.1038/sj.leu.2404258 PMid:16688224
- O'Neil
J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, Alt FW,
Kelliher M, Look AT. Activating Notch1 mutations in mouse models of
T-ALL. Blood. 2006;107(2):781-785. https://doi.org/10.1182/blood-2005-06-2553 PMid:16166587 PMCid:PMC1895623
- Deangelo
DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I, Dallob A,
Matthews C, Stone J, Freedman SJ, Aster J. A phase I clinical trial of
the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic
leukemia/lymphoma (T-ALL) and other leukemias. J Clin Onc.
2006;24(18S):6585.
- Milano
J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT,
Zacco A, Greenberg B, Ciaccio PJ. Modulation of notch processing by
gamma-secretase inhibitors causes intestinal goblet cell metaplasia and
induction of genes known to specify gut secretory lineage
differentiation. Toxicological sciences : an official journal of the
Society of Toxicology. 2004;82(1):341-358. https://doi.org/10.1093/toxsci/kfh254 PMid:15319485
- Real
PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E,
Sulis ML, Barnes K, Sawai C, Homminga I, Meijerink J, Aifantis I, Basso
G, Cordon-Cardo C, Ai W, Ferrando A. Gamma-secretase inhibitors reverse
glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat
Med. 2009;15(1):50-58. https://doi.org/10.1038/nm.1900 PMid:19098907 PMCid:PMC2692090
- Real
PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell
acute lymphoblastic leukemia. Leukemia. 2009;23(8):1374-1377. https://doi.org/10.1038/leu.2009.75 PMid:19357700 PMCid:PMC2814171
- Aster
JC, Blacklow SC. Targeting the Notch pathway: twists and turns on the
road to rational therapeutics. J Clin Oncol. 2012;30(19):2418-2420. https://doi.org/10.1200/JCO.2012.42.0992 PMid:22585704
- Cullion
K, Draheim KM, Hermance N, Tammam J, Sharma VM, Ware C, Nikov G,
Krishnamoorthy V, Majumder PK, Kelliher MA. Targeting the Notch1 and
mTOR pathways in a mouse T-ALL model. Blood. 2009;113(24):6172-6181. https://doi.org/10.1182/blood-2008-02-136762 PMid:19246562 PMCid:PMC2699237
- Extance A. Alzheimer's failure raises questions about disease-modifying strategies. Nat Rev Drug Discov. 2010;9(10):749-751. https://doi.org/10.1038/nrd3288 PMid:20885394
- Nicolas
M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H,
Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse
skin. Nature genetics. 2003;33(3):416-421. https://doi.org/10.1038/ng1099 PMid:12590261
- Demehri
S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis
by impacting the stromal microenvironment. Cancer Cell.
2009;16(1):55-66. https://doi.org/10.1016/j.ccr.2009.05.016 PMid:19573812 PMCid:PMC2705757
- Wang
NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM,
Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M,
North JP, Vemula SS, Mauro TM, Neuhaus IM, Leboit PE, Hur JS, Park K,
Huh N, Kwok PY, Arron ST, Massion PP, Bale AE, Haussler D, Cleaver JE,
Gray JW, Spellman PT, South AP, Aster JC, Blacklow SC, Cho RJ.
Loss-of-function mutations in Notch receptors in cutaneous and lung
squamous cell carcinoma. Proc Natl Acad Sci U S A.
2011;108(43):17761-17766. https://doi.org/10.1073/pnas.1114669108 PMid:22006338 PMCid:PMC3203814
- Wei
P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP,
Hosea N, Sands M, Randolph S, Smeal T. Evaluation of selective
gamma-secretase inhibitor PF- 03084014 for its antitumor efficacy and
gastrointestinal safety to guide optimal clinical trial design. Mol
Cancer Ther. 2010;9(6):1618-1628. https://doi.org/10.1158/1535-7163.MCT-10-0034 PMid:20530712
- Lopez-Guerra
M, Xargay-Torrent S, Rosich L, Montraveta A, Roldan J, Matas-Cespedes
A, Villamor N, Aymerich M, Lopez-Otin C, Perez-Galan P, Roue G, Campo
E, Colomer D. The gamma-secretase inhibitor PF-03084014 combined with
fludarabine antagonizes migration, invasion and angiogenesis in
NOTCH1-mutated CLL cells. Leukemia. 2015;29(1):96-106. https://doi.org/10.1038/leu.2014.143 PMid:24781018
- Papayannidis
C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds
JM, English PA, Ozeck M, Aster JC, Kuo F, Huang D, Lira PD, McLachlan
KR, Kern KA, Garcia-Manero G, Martinelli G. A Phase 1 study of the
novel gamma-secretase inhibitor PF-03084014 in patients with T-cell
acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood
Cancer J. 2015;5:e350. https://doi.org/10.1038/bcj.2015.80 PMid:26407235 PMCid:PMC4648526
- Zweidler-McKay
P DD, Douer D, et al. The safety and activity of BMS-906024, a gamma
secretase inhibitor (GSI) with anti-notch activity, in patients with
relapsed T-cell acute lymphoblastic leukemia (T-ALL): initial results
of a phase I trial [abstract]. Blood. 2014;121(21):Abstract 968.
- Secchiero
P, Voltan R, Rimondi E, Melloni E, Athanasakis E, Tisato V, Gallo S,
Rigolin GM, Zauli G. The gamma-secretase inhibitors enhance the
anti-leukemic activity of ibrutinib in B-CLL cells. Oncotarget.
2017;8(35):59235-59245. https://doi.org/10.18632/oncotarget.19494 PMid:28938632 PMCid:PMC5601728
- Bai
XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW, Shi Y. An
atomic structure of human gamma-secretase. Nature.
2015;525(7568):212-217. https://doi.org/10.1038/nature14892 PMid:26280335 PMCid:PMC4568306
- Hayashi
I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M,
Iwanari H, Osawa S, Morohashi Y, Li T, Wong PC, Chiba S, Kodama T,
Hamakubo T, Tomita T, Iwatsubo T. Neutralization of the gamma-secretase
activity by monoclonal antibody against extracellular domain of
nicastrin. Oncogene. 2012;31(6):787-798. https://doi.org/10.1038/onc.2011.265 PMid:21725355 PMCid:PMC4058788
- Walker
ES, Martinez M, Wang J, Goate A. Conserved residues in juxtamembrane
region of the extracellular domain of nicastrin are essential for
gamma-secretase complex formation. J Neurochem. 2006;98(1):300-309. https://doi.org/10.1111/j.1471-4159.2006.03881.x PMid:16805816
- Previs
RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational
and therapeutic implications of the Notch signaling pathway in cancer.
Clin Cancer Res. 2015;21(5):955-961. https://doi.org/10.1158/1078-0432.CCR-14-0809 PMid:25388163 PMCid:PMC4333206
- Ran
Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, Minter LM,
Osborne BA, Miele L, Golde TE. gamma-Secretase inhibitors in cancer
clinical trials are pharmacologically and functionally distinct. EMBO
Mol Med. 2017;9(7):950-966. https://doi.org/10.15252/emmm.201607265 PMid:28539479 PMCid:PMC5494507
- Palomero
T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E,
Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo
M, Nagase S, Cordon- Cardo C, Parsons R, Zuniga-Pflucker JC, Dominguez
M, Ferrando AA. Mutational loss of PTEN induces resistance to NOTCH1
inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203-1210. https://doi.org/10.1038/nm1636 PMid:17873882 PMCid:PMC2600418
- Palomero
T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in
NOTCH1-induced leukemia. Cell Cycle. 2008;7(8):965-970. https://doi.org/10.4161/cc.7.8.5753 PMid:18414037 PMCid:PMC2600414
- Knoechel
B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM,
Fernandez D, Ku M, Wang H, Piccioni F, Silver SJ, Jain M, Pearson D,
Kluk MJ, Ott CJ, Shultz LD, Brehm MA, Greiner DL, Gutierrez A,
Stegmaier K, Kung AL, Root DE, Bradner JE, Aster JC, Kelliher MA,
Bernstein BE. An epigenetic mechanism of resistance to targeted therapy
in T cell acute lymphoblastic leukemia. Nature genetics. 2014. https://doi.org/10.1158/1538-7445.AM2014-4782
- Roti
G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, Perrimon N, Pear
WS, Kung AL, Blacklow SC, Aster JC, Stegmaier K. Complementary genomic
screens identify SERCA as a therapeutic target in NOTCH1 mutated
cancer. Cancer Cell. 2013;23(3):390- 405. https://doi.org/10.1016/j.ccr.2013.01.015 PMid:23434461 PMCid:PMC3709972
- Corsello
SM, Roti G, Ross KN, Chow KT, Galinsky I, DeAngelo DJ, Stone RM, Kung
AL, Golub TR, Stegmaier K. Identification of AML1-ETO modulators by
chemical genomics. Blood. 2009;113(24):6193-6205. https://doi.org/10.1182/blood-2008-07-166090 PMid:19377049 PMCid:PMC2699238
- Roti G, Stegmaier K. New Approaches to Target T-ALL. Frontiers in oncology. 2014;4:170. https://doi.org/10.3389/fonc.2014.00170 PMid:25072021 PMCid:PMC4085879
- Roti G, Stegmaier K. Genetic and proteomic approaches to identify cancer drug targets. Br J Cancer. 2012;106(2):254-261. https://doi.org/10.1038/bjc.2011.543 PMid:22166799 PMCid:PMC3262130
- Frumm
SM, Fan ZP, Ross KN, Duvall JR, Gupta S, VerPlank L, Suh BC, Holson E,
Wagner FF, Smith WB, Paranal RM, Bassil CF, Qi J, Roti G, Kung AL,
Bradner JE, Tolliday N, Stegmaier K. Selective HDAC1/HDAC2 inhibitors
induce neuroblastoma differentiation. Chemistry & biology.
2013;20(5):713-725. https://doi.org/10.1016/j.chembiol.2013.03.020 PMid:23706636 PMCid:PMC3919449
- Banerji
V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, Kakoza RM, Chow KT,
Ross L, Alexe G, Tolliday N, Inguilizian H, Galinsky I, Stone RM,
DeAngelo DJ, Roti G, Aster JC, Hahn WC, Kung AL, Stegmaier K. The
intersection of genetic and chemical genomic screens identifies
GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest.
2012;122(3):935-947. https://doi.org/10.1172/JCI46465 PMid:22326953 PMCid:PMC3287215
- Iniguez
AB, Alexe G, Wang EJ, Roti G, Patel S, Chen L, Kitara S, Conway A,
Robichaud AL, Stolte B, Bandopadhayay P, Goodale A, Pantel S, Lee Y,
Cheff DM, Hall MD, Guha R, Davis MI, Menard M, Nasholm N, Weiss WA, Qi
J, Beroukhim R, Piccioni F, Johannessen C, Stegmaier K. Resistance to
Epigenetic-Targeted Therapy Engenders Tumor Cell Vulnerabilities
Associated with Enhancer Remodeling. Cancer Cell. 2018;34(6):922-938
e927. https://doi.org/10.1016/j.ccell.2018.11.005 PMid:30537514
- Antonello
ZA, Hsu N, Bhasin M, Roti G, Joshi M, Van Hummelen P, Ye E, Lo AS,
Karumanchi SA, Bryke CR, Nucera C. Vemurafenib-resistance via de novo
RBM genes mutations and chromosome 5 aberrations is overcome by
combined therapy with palbociclib in thyroid carcinoma with
BRAF(V600E). Oncotarget. 2017;8(49):84743- 84760. https://doi.org/10.18632/oncotarget.21262 PMid:29156680 PMCid:PMC5689570
- Place
AE, Pikman Y, Stevenson KE, Harris MH, Pauly M, Sulis ML, Hijiya N,
Gore L, Cooper TM, Loh ML, Roti G, Neuberg DS, Hunt SK, Orloff-Parry S,
Stegmaier K, Sallan SE, Silverman LB. Phase I trial of the mTOR
inhibitor everolimus in combination with multi-agent chemotherapy in
relapsed childhood acute lymphoblastic leukemia. Pediatr Blood Cancer.
2018;65(7):e27062. https://doi.org/10.1002/pbc.27062 PMid:29603593
- Aster JC, Pear WS, Blacklow SC. Notch signaling in leukemia. Annu Rev Pathol. 2008;3:587-613. https://doi.org/10.1146/annurev.pathmechdis.3.121806.154300 PMid:18039126 PMCid:PMC5934586
- Stegmaier
K, Ross KN, Colavito SA, O'Malley S, Stockwell BR, Golub TR. Gene
expression-based high-throughput screening(GE-HTS) and application to
leukemia differentiation. Nature genetics. 2004;36(3):257-263. https://doi.org/10.1038/ng1305 PMid:14770183
- Periz
G, Fortini ME. Ca(2+)-ATPase function is required for intracellular
trafficking of the Notch receptor in Drosophila. EMBO J.
1999;18(21):5983-5993. https://doi.org/10.1093/emboj/18.21.5983 PMid:10545110 PMCid:PMC1171664
- Lytton
J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or
endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem.
1991;266(26):17067-17071.
- Mahalingam
D, Cetnar J, Wilding G, Denmeade S, Sarantopoulos J, Kurman M, Carducci
M. Abstract B244: A first-in-human phase 1 clinical study of G-202, a
thapsigargin-based Prostate-Specific Membrane Antigen (PSMA) activated
prodrug, in patients with advanced solid tumors. Molecular Cancer
Therapeutics. 2013;12(11 Supplement):B244. https://doi.org/10.1158/1535-7163.TARG-13-B244
- Doan
NT, Paulsen ES, Sehgal P, Moller JV, Nissen P, Denmeade SR, Isaacs JT,
Dionne CA, Christensen SB. Targeting thapsigargin towards tumors.
Steroids. 2015;97:2-7. https://doi.org/10.1016/j.steroids.2014.07.009 PMid:25065587 PMCid:PMC4696022
- Christensen
SB, Skytte DM, Denmeade SR, Dionne C, Moller JV, Nissen P, Isaacs JT. A
Trojan horse in drug development: targeting of thapsigargins towards
prostate cancer cells. Anti-cancer agents in medicinal chemistry.
2009;9(3):276-294. https://doi.org/10.2174/1871520610909030276 PMid:19275521
- Roti
G, Qi J, Kitara S, Sanchez-Martin M, Saur Conway A, Varca AC, Su A, Wu
L, Kung AL, Ferrando AA, Bradner JE, Stegmaier K. Leukemia-specific
delivery of mutant NOTCH1 targeted therapy. The Journal of experimental
medicine. 2017. https://doi.org/10.1084/jem.20151778 PMid:29158376 PMCid:PMC5748843
- De
Ford C, Heidersdorf B, Haun F, Murillo R, Friedrich T, Borner C,
Merfort I. The clerodane diterpene casearin J induces apoptosis of
T-ALL cells through SERCA inhibition, oxidative stress, and
interference with Notch1 signaling. Cell Death Dis. 2016;7:e2070. https://doi.org/10.1038/cddis.2015.413 PMid:26821066 PMCid:PMC4816186
- Aridoss
G, Zhou B, Hermanson DL, Bleeker NP, Xing C. Structure-activity
relationship (SAR) study of ethyl
2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-
chromene-3-carboxylate (CXL017) and the potential of the lead against
multidrug resistance in cancer treatment. Journal of medicinal
chemistry. 2012;55(11):5566-5581. https://doi.org/10.1021/jm300515q PMid:22582991 PMCid:PMC6518390
- Bleeker
NP, Cornea RL, Thomas DD, Xing C. A novel SERCA inhibitor demonstrates
synergy with classic SERCA inhibitors and targets multidrug-resistant
AML. Mol Pharm. 2013;10(11):4358-4366. https://doi.org/10.1021/mp400458u PMid:24079514 PMCid:PMC3946399
- Roti
G RKN, Ferrando A.A., Blacklow S.C., Aster J, Stegmaier K.
Expression-Based Screen Identifies the Calcium Channel Antagonist
Bepridil as a Notch1 Modulator in T- ALL. . Blood. 2009;114:366.
- Baldoni
S, Del Papa B, Dorillo E, Aureli P, De Falco F, Rompietti C, Sorcini D,
Varasano E, Cecchini D, Zei T, Di Tommaso A, Rosati E, Alexe G, Roti G,
Stegmaier K, Di Ianni M, Falzetti F, Sportoletti P. Bepridil exhibits
anti-leukemic activity associated with NOTCH1 pathway inhibition in
chronic lymphocytic leukemia. Int J Cancer. 2018;143(4):958-970. https://doi.org/10.1002/ijc.31355 PMid:29508386 PMCid:PMC6055653
- Stahl
M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and
O-fucose in mammalian Notch signaling. J Biol Chem.
2008;283(20):13638-13651. https://doi.org/10.1074/jbc.M802027200 PMid:18347015 PMCid:PMC2376238
- McMillan
BJ, Zimmerman B, Egan ED, Lofgren M, Xu X, Hesser A, Blacklow SC.
Structure of human POFUT1, its requirement in ligand-independent
oncogenic Notch signaling, and functional effects of Dowling-Degos
mutations. Glycobiology. 2017;27(8):777-786. https://doi.org/10.1093/glycob/cwx020 PMid:28334865 PMCid:PMC5881682
- Koyama
D, Kikuchi J, Hiraoka N, Wada T, Kurosawa H, Chiba S, Furukawa Y.
Proteasome inhibitors exert cytotoxicity and increase chemosensitivity
via transcriptional repression of Notch1 in T-cell acute lymphoblastic
leukemia. Leukemia. 2014;28(6):1216- 1226. https://doi.org/10.1038/leu.2013.366 PMid:24301524 PMCid:PMC4051216
- Bertaina
A, Vinti L, Strocchio L, Gaspari S, Caruso R, Algeri M, Coletti V,
Gurnari C, Romano M, Cefalo MG, Girardi K, Trevisan V, Bertaina V,
Merli P, Locatelli F. The combination of bortezomib with chemotherapy
to treat relapsed/refractory acute lymphoblastic leukaemia of
childhood. Br J Haematol. 2017;176(4):629-636. https://doi.org/10.1111/bjh.14505 PMid:28116786
- Hubmann
R, Duchler M, Schnabl S, Hilgarth M, Demirtas D, Mitteregger D, Holbl
A, Vanura K, Le T, Look T, Schwarzmeier JD, Valent P, Jager U, Shehata
M. NOTCH2 links protein kinase C delta to the expression of CD23 in
chronic lymphocytic leukaemia (CLL) cells. Br J Haematol.
2010;148(6):868-878. https://doi.org/10.1111/j.1365-2141.2009.08024.x PMid:19995395
- Hubmann
R, Schwarzmeier JD, Shehata M, Hilgarth M, Duechler M, Dettke M, Berger
R. Notch2 is involved in the overexpression of CD23 in B-cell chronic
lymphocytic leukemia. Blood. 2002;99(10):3742-3747. https://doi.org/10.1182/blood.V99.10.3742 PMid:11986231
- Pahler
JC, Ruiz S, Niemer I, Calvert LR, Andreeff M, Keating M, Faderl S,
McConkey DJ. Effects of the proteasome inhibitor, bortezomib, on
apoptosis in isolated lymphocytes obtained from patients with chronic
lymphocytic leukemia. Clin Cancer Res. 2003;9(12):4570-4577.
- Masdehors
P, Merle-Beral H, Magdelenat H, Delic J. Ubiquitin-proteasome system
and increased sensitivity of B-CLL lymphocytes to apoptotic death
activation. Leuk Lymphoma. 2000;38(5-6):499-504. https://doi.org/10.3109/10428190009059268 PMid:10953970
- Duechler
M, Shehata M, Schwarzmeier JD, Hoelbl A, Hilgarth M, Hubmann R.
Induction of apoptosis by proteasome inhibitors in B-CLL cells is
associated with downregulation of CD23 and inactivation of Notch2.
Leukemia. 2005;19(2):260-267. https://doi.org/10.1038/sj.leu.2403592 PMid:15565166
- Arkwright
R, Pham TM, Zonder JA, Dou QP. The preclinical discovery and
development of bortezomib for the treatment of mantle cell lymphoma.
Expert Opin Drug Discov. 2017;12(2):225-235. https://doi.org/10.1080/17460441.2017.1268596 PMid:27917682 PMCid:PMC5520581
- Pham
LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive
NF-kappa B activation in mantle cell lymphoma B cells leads to
induction of cell cycle arrest and apoptosis. J Immunol.
2003;171(1):88-95. https://doi.org/10.4049/jimmunol.171.1.88 PMid:12816986
- Manni
S, Brancalion A, Mandato E, Tubi LQ, Colpo A, Pizzi M, Cappellesso R,
Zaffino F, Di Maggio SA, Cabrelle A, Marino F, Zambello R, Trentin L,
Adami F, Gurrieri C, Semenzato G, Piazza F. Protein kinase CK2
inhibition down modulates the NF-kappaB and STAT3 survival pathways,
enhances the cellular proteotoxic stress and synergistically boosts the
cytotoxic effect of bortezomib on multiple myeloma and mantle cell
lymphoma cells. PLoS One. 2013;8(9):e75280. https://doi.org/10.1371/journal.pone.0075280 PMid:24086494 PMCid:PMC3785505
- Weigert
O, Pastore A, Rieken M, Lang N, Hiddemann W, Dreyling M. Sequence-
dependent synergy of the proteasome inhibitor bortezomib and cytarabine
in mantle cell lymphoma. Leukemia. 2007;21(3):524-528. https://doi.org/10.1038/sj.leu.2404511 PMid:17268531
- Perez-Galan
P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome
inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through
generation of ROS and Noxa activation independent of p53 status. Blood.
2006;107(1):257-264. https://doi.org/10.1182/blood-2005-05-2091 PMid:16166592
- Weigert
O, Weidmann E, Mueck R, Bentz M, von Schilling C, Rohrberg R,
Jentsch-Ullrich K, Hiddemann W, Dreyling M. A novel regimen combining
high dose cytarabine and bortezomib has activity in multiply relapsed
and refractory mantle cell lymphoma - long- term results of a
multicenter observation study. Leuk Lymphoma. 2009;50(5):716-722. https://doi.org/10.1080/10428190902856790 PMid:19347767
- Hutter
G, Rieken M, Pastore A, Weigert O, Zimmermann Y, Weinkauf M, Hiddemann
W, Dreyling M. The proteasome inhibitor bortezomib targets cell cycle
and apoptosis and acts synergistically in a sequence-dependent way with
chemotherapeutic agents in mantle cell lymphoma. Ann Hematol.
2012;91(6):847-856. https://doi.org/10.1007/s00277-011-1377-y PMid:22231280
- Heider
U, von Metzler I, Kaiser M, Rosche M, Sterz J, Rotzer S, Rademacher J,
Jakob C, Fleissner C, Kuckelkorn U, Kloetzel PM, Sezer O. Synergistic
interaction of the histone deacetylase inhibitor SAHA with the
proteasome inhibitor bortezomib in mantle cell lymphoma. Eur J
Haematol. 2008;80(2):133-142. https://doi.org/10.1111/j.1600-0609.2007.00995.x PMid:18005386
- Qu
FL, Xia B, Li SX, Tian C, Yang HL, Li Q, Wang YF, Yu Y, Zhang YZ.
Synergistic suppression of the PI3K inhibitor CAL-101 with bortezomib
on mantle cell lymphoma growth. Cancer Biol Med. 2015;12(4):401-408.
- Alinari
L, White VL, Earl CT, Ryan TP, Johnston JS, Dalton JT, Ferketich AK,
Lai R, Lucas DM, Porcu P, Blum KA, Byrd JC, Baiocchi RA. Combination
bortezomib and rituximab treatment affects multiple survival and death
pathways to promote apoptosis in mantle cell lymphoma. MAbs.
2009;1(1):31-40. https://doi.org/10.4161/mabs.1.1.7472 PMid:20046572 PMCid:PMC2715189
- Moellering
RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL,
Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH
transcription factor complex. Nature. 2009;462(7270):182-188. https://doi.org/10.1038/nature08543 PMid:19907488 PMCid:PMC2951323
- Weber
D, Bauer M, Murone M, Lehal R, Bourquin J-P, Frismantas V, Radtke F.
411 PPharmacological activity of CB-103: An oral pan-NOTCH inhibitor
with a novel mode of action. Annals of Oncology. 2017;28(suppl_5). https://doi.org/10.1093/annonc/mdx367.044 PMid:28184416 PMCid:PMC5452071
- Spriano
F, Tarantelli C, Arribas A, Gaudio E, Cascione L, Aresu L, Zucca E,
Rossi D, Stathis A, Murone M, Weber D, Lehal R, Radtke F, Bertoni F.
Abstract B061: Targeting lymphomas with the novel first-in-class
pan-NOTCH transcription inhibitor CB-103. Molecular Cancer
Therapeutics. 2018;17:B061-B061. https://doi.org/10.1158/1535-7163.TARG-17-B061
- Garcia
JMP, Cortés J, Stathis A, Mous R, López-Miranda E, Azaro A, Genta S,
Nuciforo P, Vivancos A, Ferrarotto R, Bertoni F, Rossi D, Burr NS,
Schönborn-Kellenberger O, Jorga K, Beni L, Lehal R, Bauer M, Weber D,
Garralda E. First-in-human phase 1-2A study of CB-103, an oral
Protein-Protein Interaction Inhibitor targeting pan-NOTCH signalling in
advanced solid tumors and blood malignancies. Journal of Clinical
Oncology. 2018;36(15_suppl):TPS2619-TPS2619. https://doi.org/10.1200/JCO.2018.36.15_suppl.TPS2619
[TOP]