Giovanna Cannas, Salima Merazga and Emilie Virot.
Hospices Civils de Lyon,
Hôpital Edouard Herriot, Médecine Interne, Centre de Référence
Constitutif: Syndromes Drépanocytaires Majeurs, Thalassémies at Autres
Pathologies Rares du Globule Rouge et de l’Erythropoïèse; Lyon, France.
Correspondence to: Giovanna Cannas, M.D. Hospices Civils de Lyon;
Hôpital Edouard Herriot; Centre de Référence Constitutif: Syndromes
Drépanocytaires Majeurs, Thalassémies et Autres Pathologies Rares du
Globule Rouge et de l’Erythropoïèse; Médecine Interne, Pav.O; 5, place
d’Arsonval 69437 Lyon cedex 03, France. Tel.: +33 (0)472117413, Fax:
+33 (0)472117308. E-mail:
giovanna.cannas@chu-lyon.fr
Published: July 01, 2019
Received: March 4, 2019
Accepted: May 21, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019042 DOI
10.4084/MJHID.2019.042
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Infections,
especially pneumococcal septicemia, meningitis, and Salmonella
osteomyelitis, are a major cause of morbidity and mortality in patients
with sickle cell disease (SCD). SCD increased susceptibility to
infection, while infection leads to SCD-specific pathophysiological
changes. The risk of infectious complications is highest in children
with a palpable spleen before six months of age. Functional
splenectomy, the results of repeated splenic infarctions, appears to be
a severe host-defense defect. Infection is the leading cause of death,
particularly in less developed countries. Defective host-defense
mechanisms enhance the risk of pneumococcal complications.
Susceptibility to Salmonella infections can be explained at least in
part by a similar mechanism. In high-income countries, the efficacy of
the pneumococcal vaccine has been demonstrated in this disease. A
decreased in infection incidence has been noted in SCD patients treated
prophylactically with daily oral penicillin. Studies in low-income
countries suggest the involvement of a different spectrum of
etiological agents.
|
Introduction
Sickle
cell disease (SCD) represents an increasing global health problem. It
corresponds to an autosomal recessive disorder in which structurally
abnormal hemoglobin (HbS) leads to chronic hemolytic anemia and a
variety of severe clinical manifestations. This disorder is caused by a
point mutation. A single DNA base change leads to substitution of
valine for glutamic acid at the 6th position on beta globin chain. SCD
is one of the most common monogenic disorder.[1] SCD
is mainly widespread throughout most of the African continent, the
Middle East and India, and in localized areas in Mediterranean
countries because of a selective advantage conferred by this disorder
in protecting against Plasmodium falciparum malaria infection in heterozygotes.[2]
Because
population movements, the distribution of SCD has spread far beyond
non-endemic regions with an increase in the prevalence and genetic
heterogeneity of hemoglobinopathies across the world.[3]
The increase of inherited hemoglobin disorders will represent a severe
global health burden for the future, both in high-income and
lower-income countries.[4] In high-income countries,
this increase is in part related to significant gains in life
expectancy with a significant decrease in childhood mortality because
of better newborn screening, antibiotic prophylaxis, and hydroxyurea
therapy. Clinical outcomes have gradually improved over the years,
mostly as a result of developments in supportive care and treatment
with hydroxyurea, for many years the sole approved pharmacologic
therapy for SCD.[5] Hydroxyurea has multiple
beneficial effects for patients with SCD. Hydroxyurea causes an
increase in HbF, which interferes with the polymerization of HbS and
reduces the frequency and severity of the painful crisis.[6]
Hydroxyurea also lowers the leukocyte and platelets counts and improves
blood rheology. Vaso-occlusion typically causes acute complications,
including ischemic damage of tissues. With growing evidence of the
safety and efficacy of hydroxyurea, its use has increased in high- and
lower-income countries, but it continues to be underused.[7] Alongside hydroxyurea, novel therapeutic agents inducing HbF are currently under investigations.[8] The survival of children with SCD approaches that of unaffected children.[9]
However, this does not always apply to patients in lower-income
countries because disease management remains costly, with full access
to care only for the most privileged.[10] Life expectancy among African people with SCD is probably less than 20 years.[11]
Although over the last decade childhood mortality has been reduced,
mortality among children younger than five years remains as high as
90%.[12] Increased early mortality in Africa among children with SCD is primarily due to increased risk of infection.[13]
The lack of basic health care infrastructures often limits in most of
these countries the development of management and prevention of the
disease. Furthermore, a much more severe course of the disease is
usually observed in patients living in low-income countries compared to
genetically similar patients living in the northern hemisphere because
of environmental factors.[14]
This short review
summarizes published data regarding infections in SCD, including
interactions with environmental factors, and their specificities
according to patients living in high- or low-income countries in order
to improve patients’ care and to guide future areas for research.
Environmental Determinants SCD and Infections
Non-genetic
factors have been shown to influence the outcome of SCD. Potential
relevant environmental factors include the climate and air quality,
housing and socio-economic status, physical activities, each of which
being able to impact on SCD outcome. However, study results are
confusing and sometimes conflicting because of the complex
relationships between environmental factors and potential infections.
The rate of HbS polymerization is dependent on hypoxia, pH,
temperature, and patient’s hydration, which could be altered by
environmental factors.[15] However, inconsistencies
among studies, especially according to high- or lower-income countries,
may reflect differences in housing and social factors. Cold weather can
cause increased infections and peripheral vasoconstriction leading to
higher deoxygenation.[15] Increased blood viscosity and cold diuresis could participate in increased sickle pain in cold winter months.[16]
However, if studies conducted in both high-income countries and
lower-income countries reported a relationship between cold weather and
acute pain,[17-19] this was not confirmed by others.[20,21]
Conversely, fresh accommodation may be important in tropical countries
by protecting patients from the effects of extreme heat.[15] Similarly, higher wind speeds have been associated with increased hospital admissions for pain.[22,23] Both high and low humidity have also been associated with increased hospital admissions for pain.[22] Increased episodes of pain were reported during the rainy season under tropical climates,[17] but not in Western countries with rainy climates.[22]
Air pollution has also been reported as a leading cause of illness in
SCD. There is also evidence of a relationship between tobacco smoke and
SCD through infections, inflammation, oxidative stress and endothelial
dysfunction.[24,25] Socio-economic factors influence
the course of SCD. Increased poverty is associated with a worth outcome
in which infections may play significant part.[26]
Deficiencies in micronutrients could affect immune function and
contribute to susceptibility to infection. Suppressed cell-mediated
immunity with zinc deficiency and decreased nucleoside phosphorylase
activity has been described in SCD.[27] Giving
supplementation has been shown to increase levels of IL-2, a cytokine
needed for expansion and maintenance of T cells, and reduce the
incidence of bacterial infections.[28]
Impaired Splenic Function in SCD and Infections
The
spleen performs several essential host defense functions and plays a
key role in the increased susceptibility to certain bacterial
infections in SCD. As a phagocytic filter, it can nonspecifically
survey and present intravascular antigen to T and B cells that reside
in or transit through this lymphoid organ. The spleen is also an
important site of IgM production and memory B-cell differentiation
during primary humoral responses. It is responsible for generating
antibody responses to polysaccharide antigens. Increased susceptibility
to infections is observed in individuals undergoing splenectomy and in
those with nonfunctioning spleens. In these situations, slow flow is
created, enabling splenic macrophages to remove defective red blood
cells and bacteria and to present antigen to lymphocytes.[29] A deficient opsonization due to a defect in the alternative pathway of complement has been demonstrated.[30]
Impaired antibody formation may be the central factor responsible for
the observed serum opsonizing defects. While macrophages directly
recognize opsonized bacteria, poorly opsonized bacteria are only
cleared effectively by the spleen. Such pathogens include encapsulated
bacteria. The hyposplenic state observed in individuals with SCD is
initially reversible, then with repeated episodes of sickling and
ischemic damage spleen shrinks to a small remnant and the individual is
rendered asplenic.
Interactions Between SCD and Infections
SCD increased susceptibility to infection, while infection leads to SCD-specific pathophysiological changes (Figure 1).
SCD can create an environment supporting infections. The vast majority
of SCD patients live in low-income countries with high prevalence and
transmission rates of infections. The potential mechanisms leading red
cell sickling and vaso-occlusive crisis in SCD patients with infections
have been recently reviewed focusing on the challenging issue of
infectious diseases given the background immunodeficiency associated
with SCD and the high prevalence of infections in underdeveloped
countries.[31] Areas of necrotic bone act as foci for
infection. Salmonella is the most common agent of cases of acute
osteomyelitis in SCD (42% to 57%),[32,33] followed by Staphylococcus aureus, and then Gram-negative enteric bacteria.[34] Most of Salmonella infections were Salmonella typhimurium.[35] Infarctions of bowel secondary to microvascular occlusion favor gut bacteria to enter the bloodstream. Edwardsiella tarda is an enterobacterium that has been reported with increased incidence in SCD.[36] SCD also carries an increased risk of severe respiratory infections involving particularly Mycoplasma and Chlamydia.[37]
Reversely, infection is one of the most common factors susceptible to
induce crisis in SCD. Infection can lead to a range of complications in
SCD. During infections, changes occurring at a cellular level
predispose to crises. Circulating leukocytes and the levels of
inflammatory cytokines increase. Adhesion molecule expressions increase
on both the vascular endothelium and leukocytes. Leukocyte adhesion may
be the initiating event in vaso-occlusive episodes, as microvascular
occlusion occurs in post-capillary venules.[38] Cytotoxic proteins are produced and generate reactive O2
radicals leading to oxidative damage. The sickling process is initially
reversible when HbS is re-oxygenated, but dehydration increases HbS
concentration leading to extensive polymerization and irreversible
membrane damage. In addition, infections increase the risk of sickling
by non-specific effects through fever, anorexia, nausea, vomiting, and
diarrhea, which all contribute to dehydration.
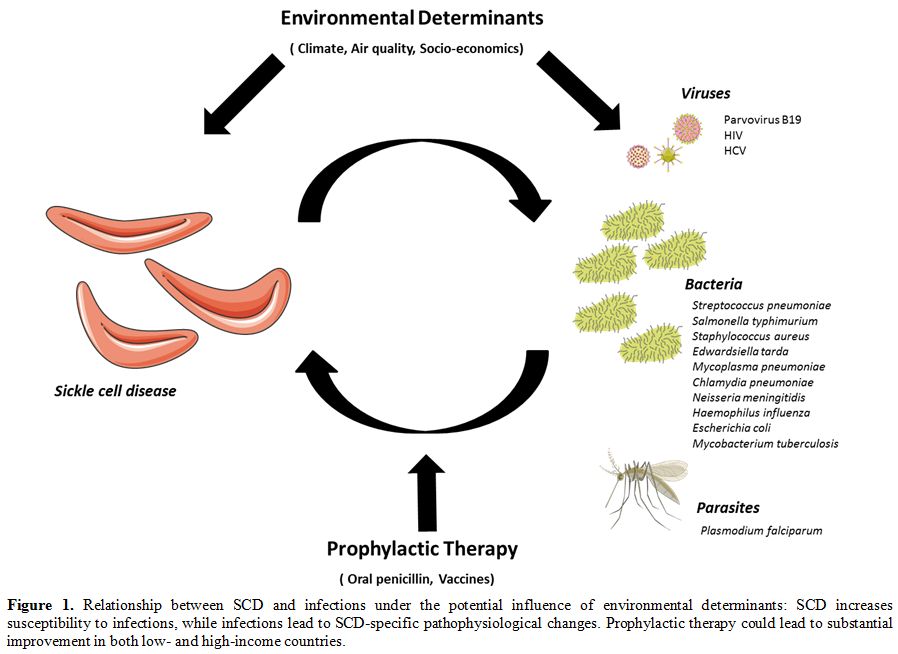 |
Figure 1. Relationship
between SCD and infections under the potential influence of
environmental determinants: SCD increases susceptibility to infections,
while infections lead to SCD-specific pathophysiological changes.
Prophylactic therapy could lead to substantial improvement in both low-
and high-income countries. |
Infections with Specific Pathogens in SCD.
Bacteria.
Local infections can become systemic. High fever is a medical emergency
in patients with SCD since it can be the first sign of bacteremia, and
a broad-spectrum parenteral antibiotic should be given without delay
after obtaining samples for blood cultures. A wide variety of organisms
have been reported to cause overwhelming sepsis, but the pneumococcus
accounts for 50-70% of such infections, with the bulk of the remainder
being accounted for Neisseria meningitidis, Haemophilus influenza, and to a lesser extent Escherichia coli.
The typical presentation is that of septic shock, disseminated
intravascular coagulopathy, and respiratory distress syndrome occurring
in the absence of a primary site of infection.[39]
Mortality can reach 35% to 50% from septicemia, and 10% in meningitis
with a risk confined almost exclusively to young children. Additional
immune deficits, including complement system deficit and reduced
leukocyte function, are present and also predispose to bacterial
infections.[40-42] These infections include Escherichia coli urinary tract infections, Mycoplasma pneumonia
respiratory infections, dental infections, and cholecystitis caused by
anaerobes. Polymorphisms of genes involved in the immune response also
contribute to increased susceptibility to infection in SCD. Particular
HLA II subtypes, polyphormisms of the FcR receptor, mannose-binding
lectin, insulin-like growth factor 1 receptor, genes from the TGFβ/
bone morphogenetic protein pathway have been involved in an increased
risk of bacteremia.[43]
Pneumococcal infections
in patients with splenectomy follow a rapidly fatal clinical course.
Disseminated intravascular coagulopathy may occur in these patients,
and organisms can be demonstrated in peripheral blood smears. The first
presentation of the disease may be sudden death due to overwhelming
sepsis. The pneumococcus is the most common cause of bacteremia and
meningitis in children with SCD. The incidence of invasive pneumococcal
disease is 300-500 times higher in SCD than in the general population
because of the loss of splenic filter function due to infarction.
Prophylactic oral penicillin reduced the risk of invasive pneumococcal
disease by 84% in children aged less than three years.[44] Fatal pneumococcal sepsis is now therefore rare in children with SCD in developed countries.[45] However, vigilance is still required because of the recent emergence of non-vaccine serotypes of Streptococcus pneumonia.[46]
Acute
chest syndrome is the second most common cause of hospital admission in
SCD and is responsible for 25% of deaths, particularly in early
childhood.[47] Infection is one of the triggers of
acute chest syndrome. Evidence of infection was found in one-third of
cases, with a demonstration of isolated pathogens or sometimes found in
combination.[38] Acute chest syndrome is common in
young children in whom it is associated with viral respiratory
infections. Acute chest syndrome could involve Chlamydia pneumoniae (14%), Mycoplasma pneumoniae (9%), and viruses in all patients with SCD regardless of age.
SCD
predisposes to osteomyelitis, which results from secondary infection of
the ischemic or avascular bone. It is often challenging to
differentiate thrombotic marrow crisis from osteomyelitis in patients
with SCD because they produce similar findings on radiographs, scans,
and magnetic resonance imaging. Clinical features are mainly a single
focus of pain, fever, and bacteremia.[48] However,
children with SCD may have multiple sites of bone infection
simultaneously. Early cultures of blood and stool offer the only clue
to the correct diagnosis. There is no standardized approach to
antibiotic therapy, and treatment is likely to vary from country to
country.[49] Presumptive antibiotic therapy should include agents effective against Salmonella.[50] Indeed, the infecting organisms were mainly gram-negative rods. Salmonella species accounted for approximately 80%. Other microorganisms included Staphylococcus aureus and Mycobacterium tuberculosis. Empiric therapy should be directed against Salmonella and Staphylococcus until an organism is identified.
Studies
on the etiological agents responsible for bacteremia in patients with
SCD in African low-income countries are few. They, however, reveal a
different spectrum of organisms than that observed in other parts of
the world. In Africa, bacteremia was found in 14% to 32% in children
with SCD. [51-54] This was much higher than the incidence observed in high-income countries.[55,56]
Reversely, to what is observed in Western countries, pneumococcal
infection in Africa does not contribute significantly to the morbidity
and mortality of children with SCD because of the involvement of other
infections, rending preventive measures inappropriate.[57] Gram-negative bacteremia constitute more than 60% of all isolates, while the predominant isolates were Klebsiella pneumonia (25%), Staphylococcus aureus (25%), and Salmonella species.[51,52,54,58-62]
One given explanation for these discrepancies in terms of patterns of
bacterial isolates was the unregulated use of antibiotics (mainly
penicillins or penicillin derivatives) before hospital admission in
some African countries, which could affect the results of bacterial
cultures.[51,63] Increased
resistance to commonly used antibiotics has been reported, but
treatment with ciprofloxacin and some third-generation cephalosporin is
still active.[61,64] Because
infections by these agents are not vaccine-preventable, it has been
suggested that disparity in terms of vaccinations among low-income and
high-income countries may not account for the higher incidence of
bacteremia in Africa, but could be explained by differences in terms of
patient’s immunity and environment.[51,65]
In Africa, patients with SCD were shown to be at increased risk of
contracting tuberculosis. They were shown to have significantly lower
hematocrit and a higher level of circulating sickle cells those
patients without tuberculosis.[66]
Viruses.
In SCD, Parvovirus B19 commonly causes a transient aplastic crisis
which occurs in 65% to 80% of infections. It specifically infects
erythroid progenitor cells resulting in a temporary cessation of
erythropoiesis leading to severe anemia.[67] Although
most children recover within two weeks, most of them require a blood
transfusion. The aplastic crisis is uncommon after 15 years old.[68]
Parvovirus aplastic crisis does not recur due to long-lasting humoral
immunity. However, infections are observed among other household
members in about 50% of cases because of the highly contagious features
of the virus.[69]
HIV prevalence in SCD patients varies between 0% and 11.5% in published studies.[70]
Few data are available regarding the impact of coexistent HIV infection
and SCD. However, this represents a challenge, particularly in Africa,
where both conditions are highest, and resources are low. Both diseases
have a common risk for stroke, splenic dysfunction, avascular necrosis,
and pulmonary arterial hypertension. HIV infection increases the risk
of sepsis and bacterial infection, mainly of pneumococcal infection.[71]
However, both diseases seem to interact closely. HIV infection tends to
decrease the risk of vaso-occlusive crisis while SCD seems to improve
the frequency of HIV long-term non-progressors.[72]
Interactions of antiretroviral therapy with SCD have been demonstrated.
A better understanding of the interactions between these diseases would
lead to better treatment approaches, especially in regions of
co-prevalence.
At least 10% of adult sickle cell patients are hepatitis C-virus (HCV) positive and often have liver dysfunction.[73]
Although the incidence of transfusion-acquired infection has decreased;
the risk is still present. The HCV antibody positivity is directly
related to the number of transfusions given.[74] Iron
overload following blood transfusions is additive to the liver damage
caused by HCV infection. The standard of care for patients with chronic
HCV infection combines interferon and ribavirin. Ribavirin (a guanosine
nucleoside analog used to treat HCV) can also increase hemolysis in
patients with SCD. In order to decrease the severity of
ribavirin-related hemolysis, it has been suggested to pre-treat HCV
patients with hydroxyurea to increase HbF.[75] Transfusions may not be the primary route of HCV transmission in lower-income countries.[76] Practices, such as circumcision and medicinal and other scarifications, may be additional risk factors.
Parasites.
The tropical environment within which most of the SCD patients live has
a very high prevalence of parasitic diseases. Malaria is a significant
pathogen in SCD. It contributes to excess mortality among patients with
SCD in Africa.[77,78] Immunological deficiencies due
to SCD render children with SCD particularly vulnerable to malaria.
Although homozygous SCD is known to confer higher resistance to
malaria, the co-existence of SCD and malaria is associated with
increased morbidity and mortality. Malaria is the most common cause of
crisis via a massive release of inflammatory cytokines. The parasite is
both erythrocytotropic and erythrocytopathic. Infected red cells sickle
as a result of metabolic changes induced by the replicating parasites
with cells becoming extremely adherent to the vascular endothelium
promoting stasis and vaso-occlusive crisis.[79] In
Africa, the tropical rainy season has been shown to be associated with
increased frequency of vaso-occlusive crisis in relationship with
increased stagnant surface waters ideal for reproduction and survival
of mosquito vectors for the malaria parasites.[79] Splenectomized individuals with Plasmodium falciparum
have reduced clearance of parasitized red blood cells and can cause
dyserythropoiesis and chronic hemolysis leading to folate-deficiency
anemia.[80] Long-term prophylaxis has been shown to lower the incidence of crisis and to reduce mortality.[81]
A
higher prevalence of protozoan and helminthic intestinal parasites in
SCD patients has been reported as a result of their weak immune
response to infection.[82] A study from Nigeria
showed that anemia in SCD patients might be exacerbated by intestinal
parasites, and suggested that these patients should have regular stool
examinations.[83] Infections were predominantly due
to soil-transmitted helminths and protozoans, strongly associated with
poverty and poor hygiene. In addition, intestinal parasites may cause
iron deficiency, which could favor cell aggregation.
Pneumonitis-induced
hypoxia and increased eosinophil counts due to tropical parasitic
diseases may increase cell adhesion to vascular endothelium
predisposing to red cell sickling and vaso-occlusive crisis.[79]
This condition includes Loffler's syndrome in ascariasis and
ancylostomiasis, schistosomiasis, filariasis, and larva migrans in
toxocariasis.
Urinary schistosomiasis is a major cause of
chronic illness endemic in Africa in both rural and urban communities
with significant socioeconomic and public health burden. A Nigerian
study showed that urinary schistosomiasis adversely affected the
severity and prognosis of SCD.[84] SCD patients with
schistosomiasis had lower hematocrit and higher reticulocyte count due
to hematuria. Higher reticulocyte, leucocyte, and thrombocyte counts
increase viscosity and accounts for the higher frequency of
vaso-occlusive crisis. Schistosomiasis was also associated with a
higher prevalence of secondary urinary tract infections including Salmonella species, Escherichia coli, Klebsiella and Staphylococcus species.
Prophylactic Therapy
Screening
programs have been established in high-income countries, and begin to
be developed in lower-income countries with a very high prevalence of
SCD. However, even if diagnostic tests can be quickly introduced in
these lower-income countries, preventive interventions not always
follow,[85] including penicillin prophylaxis in children[44] and pneumococcal vaccine.[86]
Such interventions, currently used in high-income countries, could save
millions of lives if implemented in lower-income countries.
Since
the end of the 80s, prophylactic oral penicillin V has been shown to
reduce the risk of invasive pneumococcal disease by 84% in children
aged less than three years, with minimal adverse reactions.[44,87]
This simple intervention was rapidly recommended with a beginning of
administration at 3 months in children with homozygous state for βS
(HbSS) and variants sickle-β0-thalassemia (HbSβ0)
and doses of 62.5 mg twice daily until one year, 125 mg twice daily
between one and 5 years, and 250 mg twice daily after 5 years old.[88,89]
Erythromycin is a suitable alternative in case of penicillin allergy.
For children with heterozygous state sickle-hemoglobin C disease (HbSC)
and variants sickle-β+-thalassemia (HbSβ+),
hyposplenism occurring later, practice varies among centers. However,
penicillin prophylaxis is usually considered starting at age 4-5 years
or for a history of pneumococcal sepsis or surgical splenectomy.[90]
The duration of penicillin prophylaxis remains controversial. The
absence of significant benefit has been suggested to stop prophylaxis
after five years,[89] long-term administration being a potential source of resistance development.[91] However, guidelines for asplenic patients recommend that penicillin prophylaxis be continued lifelong.[92]
Another
major key in the prevention of infection is vaccination. Early studies
with vaccination against pneumococcal bacteria suggested a 50%
reduction of invasive pneumococcal disease.[93] The
current vaccines should protect against 75% of infections, with another
14% prevented via cross-protection. For all forms of SCD, the standard
vaccine series of childhood should be considered, including the
13-valent pneumococcal conjugate vaccine. The 23-valent pneumococcal
polysaccharide vaccine should also be given at two years (and 5-yearly
after that) at least two months after the 13-valent vaccine. Other
vaccines are lifesaving in children with SCD. The 4-valent
meningococcal conjugate vaccine should be given at two years with
re-immunization considered at 5-year intervals. Annual influenza
immunization should be offered (Table 1).[89] It is expected that Salmonella vaccines may be useful in people with SCD, especially in resource-poor settings.[94] In addition, meningitidis A and C vaccination and malaria prophylaxis should be recommended for travel to endemic areas.
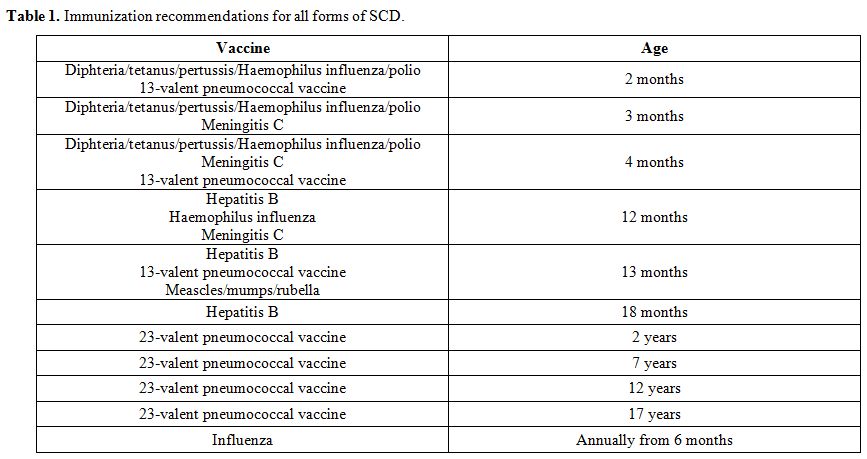 |
Table 1. Immunization recommendations for all forms of SCD. |
Conclusions
Infection
is a major determinant of the outcome in patients with SCD. It
represents the primary cause of premature deaths among children with
SCD in Africa. A substantial proportion of invasive pneumococcal and Haemophilus influenza type B disease could be attributable to SCD.[13]
The burden of SCD in Africa warrants a strong emphasis on infection
prevention, as recently stated by the World Health Organization, which
pointed to "the urgent need to develop models of care appropriate to
the management of SCD in sub-Saharan Afric".[95]
While encapsulated bacterial agents are recognized as the most
important microbes associated with severe illness, there is evidence
that SCD increases the risk for several other infections that warrant
additional preventive measures. In this setting, better identification
of risk factors could have, through the development of appropriate
public health policies, an immediate impact in preventing complications
in these patient populations. Simple measures such as better hygiene
with hand-washing, avoidance of food contamination, nutritional
supplementation can reduce infection risk.[96]
Although in a lesser extent, infections in high-income countries can
also contribute to morbidity and mortality among patients with SCD,
especially in children. However, with current multidisciplinary care,
almost all children with SCD in developed countries now survive to
adulthood. The burden of mortality has now shifted to adults. Early
identification of infections and their prompt treatment can avoid
severe complications. However, treatment of the most common bacterial
infections in SCD is not based on the results of randomized controlled
trials but based on consensus guidelines, clinical experience or
adapting treatment applied on other diseases, leading to wide
variations in treatment among institutions.[97]
Primary interventions, including penicillin prophylaxis and
vaccinations, have led to substantial improvement in higher-income
countries.[98] Recent studies showed a different
problematic in non-developed countries with a different spectrum of
organisms involved in severe infections, and highlighted the rarity of
Streptococcus pneumonia, adding to the debate regarding the need for
pneumococcal vaccines in this setting.[51]
References
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010; 376:2018-31. https://doi.org/10.1016/S0140-6736(10)61029-X
- Piel
FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell
gene and geographical confirmation of the malaria hypothesis. Nat
Commun 2010; 1:104. https://doi.org/10.1038/ncomms1104 PMid:21045822 PMCid:PMC3060623
- Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med 2017; 376:1561-1573. https://doi.org/10.1056/NEJMra1510865 PMid:28423290
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010; 115:4331-4336. https://doi.org/10.1182/blood-2010-01-251348 PMid:20233970 PMCid:PMC2881491
- Wong
TE, Brandow AM, Lim W, Lotenberg R. Update on the use of hydroxyurea
therapy in sickle cell disease. Blood 2014; 124:3850-3857. https://doi.org/10.1182/blood-2014-08-435768 PMid:25287707 PMCid:PMC4271176
- Cannas
G, Poutrel S, Thomas X. Hydroxycarbamine: from an old drug in malignant
hemopathies to a current standard in sickle cell disease. Mediterr J
Hematol Infect Dis 2017; 9:e2017015. https://doi.org/10.4084/mjhid.2017.015 PMid:28293403 PMCid:PMC5333733
- Mulaku
M, Opiyo N, Karumbi J, et al. Evidence review of hydroxyurea for the
prevention of sickle cell complications in low-income countries. Arch
Dis Child 2013; 98:908-914. https://doi.org/10.1136/archdischild-2012-302387 PMid:23995076 PMCid:PMC3812872
- Matte
A, Zorzi F, Mazzi F, et al. New therapeutic options for the treatment
of sickle cell disease. Mediterr J Hematol Infect Dis 2019;
11:e2019002. https://doi.org/10.4084/mjhid.2019.002 PMid:30671208 PMCid:PMC6328043
- Telfer
P, Coen P, Chakravorty S, et al. Clinical outcomes in children with
sickle cell disease living in England: a neonatal cohort in East
London. Haematologica 2007; 92:905-912. https://doi.org/10.3324/haematol.10937 PMid:17606440
- Kauf
TL, Coates TD, Huazhi L, et al. The cost of health care for children
and adults with sickle cell disease. Am J Hematol 2009; 84:323-327. https://doi.org/10.1002/ajh.21408 PMid:19358302
- Makani
J, Cox SE, Soka D, et al. Mortality in sickle cell anemia in Africa: a
prospective cohort study in Tanzania. PLoS One 2011; 6:e14699. https://doi.org/10.1371/journal.pone.0014699 PMid:21358818 PMCid:PMC3040170
- Grosse
SD, Odame I, Atrash HK, et al. Sickle cell disease in Africa: a
neglected cause of early childhood mortality. Am J Prev Med 2011;
41(suppl.4): S398-S405. https://doi.org/10.1016/j.amepre.2011.09.013 PMid:22099364 PMCid:PMC3708126
- Ramakrishnan
M, Moïsi JC, Klugman KP, et al. Increased risk of invasive bacterial
infections in African people with sickle-cell disease: a systematic
review and meta-analysis. Lancet 2010; 10:329-337. https://doi.org/10.1016/S1473-3099(10)70055-4
- Weatherall
MW, Higgs DR, Weiss H, et al. Phenotype/genotype relationships in
sickle cell disease: a pilot twin study. Clin Lab Haematol 2005;
27:384-390. https://doi.org/10.1111/j.1365-2257.2005.00731.x PMid:16307540
- Tewari
S, Brousse V, Piel FB, et al. Environmental determinants of severity in
sickle cell disease. Haematologica 2015; 100:1108-1116. https://doi.org/10.3324/haematol.2014.120030 PMid:26341524 PMCid:PMC4800688
- Amjad H, Bannerman RM, Judisch JM. Sickling pain and season. BMJ 1974; 2:54. https://doi.org/10.1136/bmj.2.5909.54 PMid:4821045 PMCid:PMC1610127
- Konotey-Ahulu FI. Sicklaemic human hygrometers. Lancet 1965; 1:1003-1005. https://doi.org/10.1016/S0140-6736(65)91224-9
- Smith
WR, Coyne P, Smith VS, Mercier B. Temperature changes, temperature
extremes, and their relationship to emergency department visits and
hospitalization for sickle cell crisis. Pain Manag Nurs 2003;
4:106-111. https://doi.org/10.1016/S1524-9042(02)54211-9
- Redwood
AM, Williams EM, Desal P, Serjeant GR. Climate and painful crisis of
sickle-cell disease in Jamaica. Br Med J 1976; 1:66-68.
https://doi.org/10.1136/bmj.1.6001.66 PMid:1244937 PMCid:PMC1638357
- Seeler RA. Non-seasonality of sickle-cell crisis. Lancet 1973; 2:743. https://doi.org/10.1016/S0140-6736(73)92584-1
- Slovis
CM, Talley JD, Pitts RB. Non relationship of climatologic factors and
painful sickle cell anemia crisis. J Chronic Dis 1986; 39:121-126. https://doi.org/10.1016/0021-9681(86)90068-8
- Jones
S, Duncan ER, Thomas N, et al. Windy weather and low humidity are
associated with an increased number of hospital admissions for acute
pain and sickle cell disease in an urban environment with a maritime
temperature climate. Br J Haematol 2005; 131:530-533. https://doi.org/10.1111/j.1365-2141.2005.05799.x PMid:16281945
- Nolan
VG, Zhang Y, Lash T, et al. Association between wind speed and the
occurrence of sickle cell acute painful episodes: results of a
case-crossover study. Br J Haematol 2008; 143:433-438. https://doi.org/10.1111/j.1365-2141.2008.07354.x PMid:18729854 PMCid:PMC4347894
- Cohen
RT, DeBaun MR, Blinder MA, et al. Smoking is associated with an
increased risk of acute chest syndrome and pain among adults with
sickle cell disease. Blood 2010; 115:3852-3854. https://doi.org/10.1182/blood-2010-01-265819 PMid:20448118 PMCid:PMC2915907
- West
DC, Romano PS, Azari R, et al. Impact of environmental tobacco smoke on
children with sickle cell disease. Arch Pediatr Adolesc Med 2003;
157:1197-1201. https://doi.org/10.1001/archpedi.157.12.1197 PMid:14662575
- Farber
MD, Koshy M, Kinney TR. Cooperative study of sickle cell disease:
demographic and socioeconomic characteristics of patients and families
with sickle cell disease. J Chronic Dis 1985; 38:495-505. https://doi.org/10.1016/0021-9681(85)90033-5
- Ballester
OF, Pasad AS. Anergy, zinc deficiency and decreased nucleoside
phosphorylase activity in patients with sickle cell anemia. Ann Intern
Med 1983; 98:180-182. https://doi.org/10.7326/0003-4819-98-2-180
- Prasad
AS, Back FW, Kaplan J, et al. Effect of zinc supplementation on
incidence of infections and hospital admissions in sickle cell disease.
Am J Hematol 1999; 61:194-202. https://doi.org/10.1002/(SICI)1096-8652(199907)61:3<194::AID-AJH6>3.0.CO;2-C
- Bohnsack JF, Brown EJ. The role of the spleen in resistance to infection. Ann Rev Med 1986; 37:49-59. https://doi.org/10.1146/annurev.me.37.020186.000405 PMid:3518612
- Johnston
RB Jr, Newman LS, Struth AG. An abnormality of the alternate pathway of
complement activity in sickle cell disease. N Engl J Med 1973;
288:803-808. https://doi.org/10.1056/NEJM197304192881601 PMid:4144343
- Ahmed
SG. The role of infection in the pathogenesis of vaso-occlusive crisis
in patients with sickle cell disease. Mediterr J Hematol Infect Dis
2011; 3:e2011028. https://doi.org/10.4084/mjhid.2011.028 PMid:21869914 PMCid:PMC3152450
- Chambers
JB, Forsythe DA, Bertrand SL, et al. Retrospective review of
osteoarticular infections in a pediatric sickle cell age group. J
Pediatr Orthop 2000; 20:682-685. https://doi.org/10.1097/01241398-200009000-00025 PMid:11008753
- Sadat-Ali M. The status of acute osteomyelitis in sickle cell disease. A 15-year review. Int Surg 1998; 83:84-87.
- Almeira A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol 2005; 129:482-490. https://doi.org/10.1111/j.1365-2141.2005.05476.x PMid:15877730
- Workman
MR, Philpott-Howard J, Bellingham AJ. Salmonella bacteraemia in sickle
cell disease at King's College Hospital: 1976-1991. J Hosp Infect 1994;
34:195-199. https://doi.org/10.1016/0195-6701(94)90127-9
- Wang
IK, Kuo HL, Chen YM, et al. Extraintestinal manifestations of
Edwardsiella tarda infection. Int J Clin Pract 2005; 59:917-921. https://doi.org/10.1111/j.1742-1241.2005.00527.x PMid:16033613
- Vichinsky
EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute
chest syndrome in sickle cell disease. National Acute Chest Syndrome
Study Group. N Engl J Med 2000; 342:1855-1865. https://doi.org/10.1056/NEJM200006223422502 PMid:10861320
- Manwani
D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology
and novel targeted therapies. Blood 2013; 122:3892-3898. https://doi.org/10.1182/blood-2013-05-498311 PMid:24052549 PMCid:PMC3854110
- William
BM, Thawani N, Sae-Tia S, Corazza GR. Hyposplenism: a comprehensive
review. Part II: Clinical manifestations, diagnosis and management.
Haematology 2007; 12:89-98. https://doi.org/10.1080/10245330600938463 PMid:17454188
- Tamouza
R, Neonato MG, Busson M, et al. Infectious complications in sickle cell
disease are influenced by HLA class II alleles. Hum Immunol 2002;
63:194-199. https://doi.org/10.1016/S0198-8859(01)00378-0
- Larcher
VF, Wyke RJ. Defective yeast opsonisation and functional deficiency of
complement in sickle cell disease. Arch Dis Child 1982; 57:343-346. https://doi.org/10.1136/adc.57.5.343 PMid:7092289 PMCid:PMC1627548
- Anyaegbu
CC, Okpala IE. Peripheral blood neutrophil count and candidacidal
activity correlate with the clinical severity of sickle cell anaemia.
Eur J Haematol 1998; 60:267-268. https://doi.org/10.1111/j.1600-0609.1998.tb01036.x
- Adewoye
A, Nolan V, Ma Q, et al. Association of polymorphisms of IGF1R and
genes in the TGFβ/BMP pathway with baxteremia in sickle cell anemia.
Clin Infect Dis 2006; 43:593-598. https://doi.org/10.1086/506356 PMid:16886151
- Gaston
MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in
children with sickle cell anemia - a randomized trial. N Engl J Med
1986; 314:1593-1599. https://doi.org/10.1056/NEJM198606193142501 PMid:3086721
- Quinn
CT, Rogers ZR, McCavit TL, et al. Improved survival of children and
adolescents with sickle cell disease. Blood 2010; 115:3447-3452. https://doi.org/10.1182/blood-2009-07-233700 PMid:20194891 PMCid:PMC2867259
- McCavit
TL, Quinn CT, Techasaensiri C, et al. Increase in invasive
Streptococcus pneumoniae infections in children with sickle cell
disease since pneumococcal conjugate vaccine licensure. J Pediatr 2011;
158:505-507. https://doi.org/10.1016/j.jpeds.2010.11.025 PMid:21193205 PMCid:PMC3062091
- Castro
O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in
sickle cell disease: incidence and risk factors. The cooperative study
of sickle cell disease. Blood 1994; 84:643-649.
- Berger
E, Saunders N, Wang L, et al. Sickle cell disease in children:
differentiating osteomyelitis from vaso-occlusive crisis. Arch Pediatr
Adolesc Med 2009; 163:251-255. https://doi.org/10.1001/archpediatrics.2008.545 PMid:19255393
- Marti-Carvajal
AJ, Agreda-Perez LH. Antibiotics for treating osteomyelitis in people
with sickle cell disease. Cochrane Database Syst Rev 2016; 11:CD007175.
https://doi.org/10.1002/14651858.CD007175.pub4
- Hand WL, King NL. Serum opsonization of Salmonella in sickle cell anemia. Am J Med 1978; 64:388-395. https://doi.org/10.1016/0002-9343(78)90217-6
- Brown
B, Dada-Adegbola H, Trippe C, Olopade O. Prevalence and etiology of
bacteremia in febrile children with sickle cell disease at a Nigeria
tertiary hospital. Mediterr J Hematol Infect Dis 2017; 9:e2017039. https://doi.org/10.4084/mjhid.2017.039 PMid:28698782 PMCid:PMC5499496
- Okuonghae
HO, Nwankwo MU, Offor EC. Pattern of bacteraemia in febrile children
with sickle cell anaemia.Ann Trop Paediatr 1993; 13:55-64. https://doi.org/10.1080/02724936.1993.11747625 PMid:7681646
- Brown
BJ, Jacob NE, Lagunju IA, Jarrett OO. Morbidity and mortality pattern
in hospitalized children with sickle cell disorders at the University
College Hospital, Ibadan, Nigeria. Nig J Paediatr 2013; 40:34-39. https://doi.org/10.4314/njp.v40i1.6
- Kizito
ME, Mworozi E, Ndugwa C, Serjeant GR. Bacteraemia in homozygous sickle
cell disease in Africa: is pneumococcal prophylaxis justified? Arch Dis
Child 2007; 92:21-23. https://doi.org/10.1136/adc.2005.088807 PMid:16531454 PMCid:PMC2083172
- Bansil
NH, Kim TY, Tieu L, Barcega B. Incidence of serious bacterial
infections in febrile children with sickle cell disease. Clin Pediatr
2013; 52:661-666. https://doi.org/10.1177/0009922813488645 PMid:23661790
- Morrissey
BJ, Bycroft TP, Almossawi O, et al. Incidence and predictors of
bacterial infection in febrile children with sickle cell disease.
Hemoglobin 2015; 39:316-319.
- Serjant G. Mortality from sickle cell disease in Africa. BMJ 2005; 330:432-433. https://doi.org/10.1136/bmj.330.7489.432 PMid:15731125 PMCid:PMC549643
- Akinyanju O, Johnson AO. Acute illness in Nigerian children with sickle cell anaemia. Ann Trop Paediatr 1987; 7:181-186. https://doi.org/10.1080/02724936.1987.11748503 PMid:2445266
- Akuse
RM. Variation in the pattern of bacterial infection in patients with
sickle cell disease requiring admission. J Trop Paediatr 1996;
42:318-323. https://doi.org/10.1093/tropej/42.6.318 PMid:9009554
- Makani
J, Mgaya J, Balandya E, et al. Bacteraemia in sickle cell anaemia is
associated with low haemoglobin: a report of 890 admissions to a
tertiary hospital in Tanzania. Br J Haematol 2015; 171:273. https://doi.org/10.1111/bjh.13553 PMid:26084722 PMCid:PMC4744759
- Bello
N, Kudu ATD, Adetokun AB, et al. Characterization and antimicrobial
susceptibility profile of bacteraemia causing pathogens isolated from
febrile children with and without sickle cell disease in Kano, Nigeria.
Mediterr J Hematol Infect Dis 2018; 10:e2018016. https://doi.org/10.4084/mjhid.2018.016 PMid:29531653 PMCid:PMC5841934
- Yanda
ANA, Nansseu JRN, Awa HDM, et al. Burden and spectrum of bacterial
infections among sickle cell disease children living in Cameroon. BMC
Infec Dis 2017; 17:221. https://doi.org/10.1186/s12879-017-2317-9 PMid:28298206 PMCid:PMC5353947
- Kateete
DP, Kajumbula H, Kaddu-Mulindwa DH, et al. Nasopharyngeal carriage rate
of Streptococcus pneumonia in Ugandan children with sickle cell
disease. BMC Res Notes 2012; 5:28. https://doi.org/10.1186/1756-0500-5-28 PMid:22243524 PMCid:PMC3283489
- Mava
Y, Bello M, Ambe JP, Zailani SB. Antimicrobiol sensitivity pattern of
organisms causing urinary tract infection in children with sickle cell
anemia in Maiduguri, Nigeria. J Clin Prac 2012; 15:420-423. https://doi.org/10.4103/1119-3077.104515 PMid:23238191
- Athale
UH, Chintu C. Clinical analysis of mortality in hospitalized Zambian
children with sickle cell anaemia. East Afr Med J 1994; 71:388-391.
- Ahmed
SG, Bukar AA, Jolayemi B. Hematological indices of sickle cell anaemia
patients with pulmonary tuberculosis in northern Nigeria. Mediterr J
Hematol Infect Dis 2010; 2:e20100. https://doi.org/10.4084/mjhid.2010.014 PMid:21415951 PMCid:PMC3033109
- Smith-Whitley
K, Zhao H, Hodinka RL, et al. Epidemiology of human parvovirus B19 in
children with sickle cell disease. Blood 2004; 103:422-427. https://doi.org/10.1182/blood-2003-01-0069 PMid:14525777
- Serjeant
BE, Hambleton IR. Haematological response to parvovirus B19 infection
in homozygous sickle cell disease. Lancet 2001; 358:1779-1780. https://doi.org/10.1016/S0140-6736(01)06807-6
- Servey JT, Reamy BV, Hodge J. Clinical presentations of parvovirus B19 infection. Am Fam Physician 2007; 75:373-376.
- Owusu
ED, Visser BJ, Nagel IM, et al. The interaction between sickle cell
disease and HIV infection: a systematic review. Clin Infect Dis 2015;
60:612-626. https://doi.org/10.1093/cid/ciu832 PMid:25344542
- Godeau
B, Bachir D, Schaeffer A, et al. Severe pneumococcal sepsis and
meningitis in HIV-infected adults with sickle cell disease. Clin Infect
Dis 1992; 15:327-329. https://doi.org/10.1093/clinids/15.2.327 PMid:1520768
- Bagasra
O, Steiner RM, Ballas SK, et al. Viral burden and disease progression
in HIV-1-infected patients with sickle cell anemia. Am J Hematol 1998;
59:199-207. https://doi.org/10.1002/(SICI)1096-8652(199811)59:3<199::AID-AJH4>3.0.CO;2-L
- Hasan MF, Marsh F, Posner G. Chronic hepatitis C in patients with sickle cell anemia. A J Gastroenterology 1996; 91:1204-1206.
- Hassan M, Hasan S, Castro O, et al. HCV in sickle cell disease. J Natl Med Assoc 2003; 95:864-874.
- Swaim MW, Agarwak S, Rosse W. Successful treatment of hepatitis C in sickle-cell disease. Ann Intern Med 2000; 133:750-751. https://doi.org/10.7326/0003-4819-133-9-200011070-00033 PMid:11074924
- Adewuyi
JO. Prevalence of antibodies to hepatitis C virus among normal blood
donors and multi-transfused sickle cell anemic patients in Nigeria.
Tropical Doctor 1996; 26:29-30. https://doi.org/10.1177/004947559602600111 PMid:8693560
- Makani
J, Komba AN, Cox SE, et al. Malaria in patients with sickle cell
anemia: burden, risk factors, and outcome at the outpatient clinic and
during hospitalization. Blood 2010; 115:215-220. https://doi.org/10.1182/blood-2009-07-233528 PMid:19901265 PMCid:PMC2843825
- McAuley
CF, Webb C, Makani J, et al. High mortality from Plasmodium falciparum
malaria in children living with sickle cell anemia on the coast of
Kenya. Blood 2010; 116:1663-1668. https://doi.org/10.1182/blood-2010-01-265249 PMid:20530796 PMCid:PMC3073423
- Ahmed
SG, Ibrahim UA. A compendium of pathophysiologic basis of etiologic
risk factors for painful vaso-occlusive crisis in sickle cell disease.
Niger J Basic Clin Sci 2017; 14:57-77. https://doi.org/10.4103/njbcs.njbcs_11_17
- Engwerda CR, Beattie L, Amante FH. The importance of the spleen in malaria. Trends Parasitol 2005; 21:75-80. https://doi.org/10.1016/j.pt.2004.11.008 PMid:15664530
- Athuman
M, Kabanywanyi AM, Rohwer AC. Intermittent preventive antimalarial
treatment for children with anaemia. Cochrane Database Syst Rev 2015;
1:CD010767. https://doi.org/10.1002/14651858.CD010767.pub2 PMid:25582096 PMCid:PMC4447115
- Mahdi
NK, Ali NH. Intestinal parasites, including Cryptosporidium species, in
Iraqi patients with sickle cell anaemia. Eastern Mediterr Health 2002;
8:345-349.
- Ahmed
SG, Uraka J. Impact of intestinal parasites on haematological
parameters of sickle-cell anaemia patients in Nigeria. Eastern Mediterr
Health J 2011; 17:710-713. https://doi.org/10.26719/2011.17.9.710
- Ahmed
SG, Kagu MB, Ibrahim UA. Impact of urinary schistosomiasis on
haematological parameters and frequency of vaso-occlusive crisis among
patients with sickle cell disease in northern Nigeria. Egyptian J
Haematol 2014; 39:58-63. https://doi.org/10.4103/1110-1067.139762
- Kanter
J, Telen MJ, Hoppe C, et al. Validation of a novel point of care
testing device for sickle cell disease. BMC Med 2015; 13:225. https://doi.org/10.1186/s12916-015-0473-6 PMid:26377572 PMCid:PMC4573998
- Broome
CV, Facklam RR, Fraser DW. Pneumococcal disease after pneumococcal
vaccination.An alternative method to estimate the efficacy of
pneumococcal vaccine. N Engl J Med 1980; 303:549-552. https://doi.org/10.1056/NEJM198009043031003 PMid:6995835
- Rankine-Mullings
AE, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal
infection in children with sickle cell disease. Cochrane Database Syst
Rev 2017; 10:CD003427. https://doi.org/10.1002/14651858.CD003427.pub4 PMid:28994899 PMCid:PMC6485662
- Sickle cell disease in childhood. Standards and guidelines for clinical care. Public Health England; 2010 October. https://www.gov.uk/government/publications/sickle-cell-disease-in-children-standars-for-clinical-care.
- Quinn
CT. Sickle cell disease in childhood: From newborn screening through
transition to adult medical care. Pediatr Clin North Am 2013;
60:1363-1381. https://doi.org/10.1016/j.pcl.2013.09.006 PMid:24237976 PMCid:PMC4262831
- Falletta
JM, Woods GM, Verter JI, et al. Discontinuing penicillin prophylaxis in
children with sickle cell anemia. Prophylactic penicillin study II. J
Pediatr 1995; 127:685-690. https://doi.org/10.1016/S0022-3476(95)70154-0
- Reynolds
R, Potz N, Colman M, et al. BSAC Extended Working Party on Bacteraemia
Resistance Surveillance. Antimicrobial sensitivity of the pathogens of
bacteraemia in the UK and Ireland 2001-2: the BSAC Bacteraemia
Resistance Surveillance Programme. J Antimicrob Chemother 2004;
53:1018-1032. https://doi.org/10.1093/jac/dkh232 PMid:15128723
- Davies
JM, Barnes R, Milligan D. Update of guidelines for the prevention and
treatment of infection in patients with an absent or dysfunctional
spleen. Clin Med 2002; 2:440-443. https://doi.org/10.7861/clinmedicine.2-5-440 PMid:12448592 PMCid:PMC4953085
- Overturf
GD. Prevention of invasive pneumococcal infection in sickle cell
disease: on the threshold of a new era of success? J Paediatr 2003;
143:438-444. https://doi.org/10.1067/S0022-3476(03)00466-9
- Odey
F, Okomo U, Oyo-Ita A. Vaccines for preventing invasive salmonella
infections in people with sickle cell disease. Cochrane Database Syst
Rev 2018; 12:CD006975. https://doi.org/10.1002/14651858.CD006975.pub4
- WHO. Sickle cell anemia: report by the Secretariat. Geneva: World Health Organization, 2006. http://apps.who.int/gb/ebwha/pdf_files/WHA59/A59_9-en.pdf.
- Obaro
SK, Iroh Tam PY. Preventing infections in sickle cell disease: the
unfinished business. Pediatr Blood Cancer 2016; 63:781-785. https://doi.org/10.1002/pbc.25911 PMid:26840500
- Sobota
A, Sabharwal V, Fonebi G, Steinberg M. How we prevent and manage
infection in sickle cell disease. Br J Haematol 2015; 170:757-767. https://doi.org/10.1111/bjh.13526 PMid:26018640
- Williams
TN, Uyoga S, Macharia A, et al. Bacteraemia in Kenyan children with
sickle-cell anaemia: a retrospective cohort and case-control study.
Lancet 2009; 374:1364-1370. https://doi.org/10.1016/S0140-6736(09)61374-X
[TOP]