Narjes Shokatpour3, Maryam Vaezjalali1,2, Graham R. Foster4 and Shahnaz Sali1.
1 Infectious diseases and tropical medicine research center, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2 Department of Microbiology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3 Department of Microbiology, Faculty of Biological Sciences, Shahid Beheshti University, Tehran, Iran.
4 Queen Mary, University of London, The liver unit, London, UK.
Correspondence to: Shahnaz Sali, MD. Address: Shahid Beheshti
University of Medical Sciences, Velenjak Street, Tehran, Iran. Postal
Code: 1985717443. Tel: +98 2122439965, Fax: +98 2122439964. E-mail:
dr.ShSali@gmail.com
Published: July 1 , 2019
Received: October 25, 2018
Accepted: June 10, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019046 DOI
10.4084/MJHID.2019.046
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Mutations in the S gene (HBsAg), pre-core (PC), and basic core promoter
(BCP) of the hepatitis B virus (HBV) infection are correlated with
disease progression. This study assessed the frequency of mutations in
the S gene, PC, and BCP regions in chronic hepatitis B (CHB) patients.
Methods:
104 formerly known CHB patients who visited Tehran Hepatitis centers,
were included. The viral load of samples was determined based on the
TaqMan method. Regions of the S gene, PC and BCP were amplified by the
nested PCR. Positive PCR products were sequenced and analyzed.
Results:
33 successfully sequenced S gene region revealed all the derived
strains were genotype D, with the majority (90.9%) belonging to the
ayw2 subtype, and the rest (9.1%) to the ayw1 subtype. The prevalence
of mutations was found to be 51.0% and 18.0% in the HBsAg and the Major
Hydrophilic Region, respectively. 70.0% of amino acid changes within
HBsAg occurred in different immune epitopes, of which 27.0% and 72.0%
were located in B cell and Th epitopes, respectively. 26 successfully
sequenced PC and BCP regions showed at least one mutation in 84.6% of
the HBV strains. The PC and BCP mutations were G1896A (61.0%), G1899A
(23.0%), A1762T/G1764A (23.0%) and G1764T/C1766G (26.0%). None of the
strains with A1762T/G1764A mutation carried the G1764T/C1766G mutant.
Conclusions:
Our results showed common mutations within HBsAg, occurring in immune
epitopes, a high rate of G1896A mutations in the PC region, and a
negative correlation between the emergence of A1762T/G1764A mutation
and the G1764T/C1766G mutant in the BCP region.
|
Introduction
Hepatitis
B virus infection is a global epidemic health problem, which leads to
cirrhosis, hepatocellular carcinoma, and severe liver disease.[1] Over 257 million people are chronic carriers of this virus.[1,2] The disease progression and the treatment response depend on the virus genotype in infected individuals.[3]
HBV consists of four overlapping open reading frames (ORFs) and encodes
seven viral proteins, including three envelop proteins (pre-S1, pre-S2,
S), core protein, polymerase, HBx protein (as a transcriptional
transactivator) and HBe protein.[4] HBV genome is classified into ten genotypes (from A-J) by an intergroup difference > 8%. [5]
The subtypes are associated with Hepatitis B surface antigen (HBsAg)
epitopes, located in a region that comprises the two external loops of
the molecule. HBsAg subtypes are differentiated based on two sets of
determinants (d/y and w/r) and a determinant named (q).[4]
As a result of the reverse transcriptase activity of HBV polymerase,
this virus displays a wide genetic diversity. The immune system of the
host and antiviral therapy are involved in the evolution of the HBV
genome.[6]
HBsAg is a significant target for immune-mediated virus elimination.[7]
This protein bears an antigenic structure, termed the Major Hydrophilic
Region (MHR, aa 99-169). The MHR being the main target for neutralizing
antibodies, encompasses the major B cell epitopes, the "a determinant"
domain (aa 124-147).[8] The humoral response against
HBsAg, is furthermore, T cell dependent. As a result, the appropriate
reactivity of T cell, since it was a prerequisite for adequate anti-HBs
products should affect the T cell epitopes within HBsAg which are a
target for the recognition by T cells.[7] Mutations
occurring within these immune epitopes play a significant role, and
viruses carrying such mutations are predicted to evade host immune
surveillance.[9]
Hepatitis B e antigen (HBeAg) is
a secreted protein and a marker of active viral replication. In the
natural course of infection with HBV, seroconversion from HBeAg to
anti-HBe usually indicates the end of the active viral replication.
However, mutations in the pre-core (PC) and basic core promoter (BCP)
cause HBeAg to turn negative in patients with chronic HBV, even though
replication continues and HBV-DNA is detectable.[2,10] The most common mutations involve G1896A in the PC region and the simultaneous presence of G1764A/A1762T in the BCP region,[11,12] which results to the premature termination of HBeAg expression and decreased level of HBeAg, respectively.[10-12]
Mutations
in the core/pre-core and surface regions are correlated with disease
progression, ranging from asymptomatic HBV carriers to fulminant
hepatitis. Extensive studies have been done to associate these
mutations with enhanced virulence. Nevertheless, it is still difficult
to analyze the role of viral versus host factors in the progression of
the disease.[11] In spite of extensive studies of HBV
in Iran, there is little data on hepatitis B genome characterization.
The aim of the present study was to investigate the frequency of
mutations in the core/pre-core and the surface region of the hepatitis
B virus derived from Iranian patients.
Patients and Methods
Samples.
The present study involved a total of 104 formerly known chronic
hepatitis B patients (Positive for HBsAg for at least six months) who
visited Tehran Hepatitis centers between 2014 and 2015. The ELISA
method (Biokit, Spain) was used to test HBsAg, HBsAb, HBeAg, and HBeAb
of all the serum samples. The HBsAg positive samples were subjected to
DNA extraction using a commercial kit (High pure viral nucleic acid
kit, Roche, Germany). Extracted DNA was stored at -70°C for PCR.
HBV viral load determination. The viral load of serum samples was determined by COBAS® AmpliPrep/COBAS® TaqMan® HBV Test, v2.0 (Roche, USA).
DNA amplification of the PC and BCP regions. The region harboring the pre-core and basal core promoters of HBV was amplified by nested PCR using appropriate primers.[13]
First-round PCR was carried out with 5 µl of extracted DNA in a total
amplification mixture of 50 µl containing Taq polymerase, dNTP,
primers, and PCR buffer. The PCR profile was preheated at 95°C for 15
min, followed by 35 cycles of amplification (95°C for 45 s, 53°C for 45
s, 72°C for 1 min), with a final extension at 72°C for 7 min. For the
second round PCR, 5 µl of the first round PCR products were used as a
template with the same condition as the first round.
DNA amplification of surface gene. The surface region was amplified by nested PCR using suitable primers.[14]
The PCR condition was an initial 5 min preheating at 94°C, then 35
cycle amplification (94° 30 s, 56° 30 s, 72° 60 s ) with a
final extension at 72°C for 10 min. For the second round PCR, 5 µl of
the first round PCR products were used as a template with the same
condition as the first round except for the slightly altered annealing
temperature (62°C instead of 56°C).
Sequencing and phylogenetic analysis.
The positive PCR products were subjected to purification and sequencing
(ABI 3730XL DNA Analyzer, Bioneer, Korea). Nucleotide sequences were
aligned and analyzed using Bio Edit software version 7.0.0.
Statistical analysis.
Statistical analysis was performed with Chi-square or Fisher exact test
for categorical variables and with independent samples t-test for
continuous variables using the SPSS version 21.0 software package. P
values (two-tailed) less than 0.05 were considered statistically
significant. The logarithms of HBV DNA levels were used for analysis.
This
study was approved by the ethics committee of Shahid Beheshti
University of Medical Sciences, Infectious Diseases, and Tropical
Medicine Research Center (approval number 2013/6/2), and was in
accordance with the Helsinki Declaration of 1964.
Results
Overall,
104 serum samples from patients with chronic HBV infection (75 males,
29 females; mean age 41.9+14.7 years) were obtained. A total of 20
(19.2%) patients had inactive HBV, 74 (71.2%) had chronic active HBV,
and 10 (9.6%) had cirrhosis. Patients with chronic active HBV 74/104
(71.2%) and patients with cirrhosis 10/104 (9.6%) received antiviral
therapy. Patients with a history of Lamivudine treatment were
administered with Lamivudine again, and patients with a history of
Tenofovir treatment or naïve treatment experience received Tenofovir.
55.8% (58/104) of them had a record of HBV infection among their
family. The mean AST and ALT values of patients were 107± 69.3 and
137±93.7, respectively (Table 1).
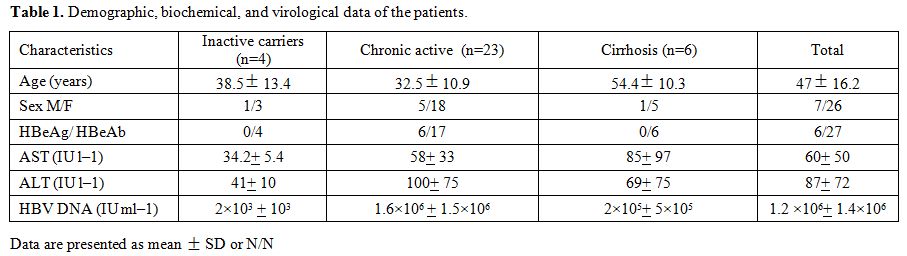 |
Table 1. Demographic, biochemical, and virological data of the patients. |
All the samples were
positive and negative for HBsAg and HBsAb, respectively. A total of
32.7% (34/104) and 66.3% (69/104) of patients had HBeAg and HBeAb,
respectively. Based on the nested PCR results, 43 (41.0%) samples were
positive for HBVDNA (limit of detection; LOD = 15 IU/mL). The surface
region could be sequenced for 33 (76.7%) HBV infecting strain, and the
sequence of the PC and BCP regions could be determined in 26 (60.5%)
cases. All the derived strains were genotype D, and the S gene
sequences revealed that the majority of isolates (30/33, 90.9%) were
found belonging to ayw2, and the rest (3/33, 9.1%) to ayw1.
Amino acid mutations within the surface gene.
Five (5) mutations were detected within the surface gene in the
patients (F8L, T118M, E164D, T189I, and W196L). The prevalence of
strains with S region mutation (single or multiple) found in all cases
was 51.0% (17/33). Eight (8) of the cases had a single mutation, 3
cases 2 mutations, 6 cases 3 mutations. The frequency of mutations in
the MHR was seen in 18.0% (6/33) of isolates (T118M, E164D). The most
common amino acid change found within HBsAg was W196L in 13 (39.0%)
isolates. A total of 32 amino acid changes, 22 (70.0%) occurred in
different immune epitopes within the surface protein, of which 6
(27.0%) and 16 (72.0%) were located in B cell and Th epitopes,
respectively (Figure 1). Also, AST was significantly higher among patients with F8L HBV mutants (P=0.001) (Table 3).
Mutations in the PC and BCP regions.
At least one mutation was detected in the PC region in 60.0% (13/22)
and 75.0% (3/4) of the HBeAg negative and HBeAg positive patients,
respectively. In the BCP region, at least one mutation was observed in
54.5% (12/22) and 75.0% (3/4) of the HBeAg negative and HBeAg positive
patients, respectively.
A high proportion (61.0%, 16/26) of
G1896A mutation occurred in the PC region. The G1899A mutation was
found in 6 (23.0%) isolates and had concomitant G1896A change. In the
BCP region, the most common mutations were A1762T (30.0%, 8/26) and
G1764T (30.0%, 8/26), followed by G1764A (26.0%, 7/26), C1766G (26.0%,
7/26), C1766T (11.5%, 3/26). A1762T and G1764A were frequently
detected together in 23.0% (6/26) of the isolates. Similarly, G1764T
and C1766G were frequently seen together in 26.0% (7/26) of cases.
However, none of the patients with A1762T/G1764A mutation carried the
G1764T/C1766G mutant (Table 2).
There was no significant relationship between BCP, pre-core and surface
mutations with HBV viral load, HBeAg and liver enzymes (Table 4), except an association between C1766G and HBeAg negativity which was significant (P=0.04).
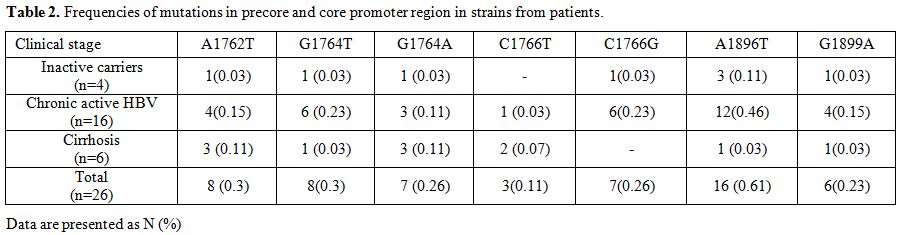 |
Table 2. Frequencies of mutations in precore and core promoter region in strains from patients. |
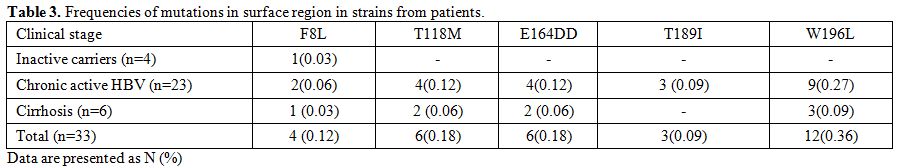 |
Table 3. Frequencies of mutations in surface region in strains from patients. |
 |
Figure 1. Amino acid mutations within B
cell, T helper (Th) and CTL epitopes of HBs proteins. Amino acids are
designated by single letter code and numbered from the beginning of
HBsAg.
|
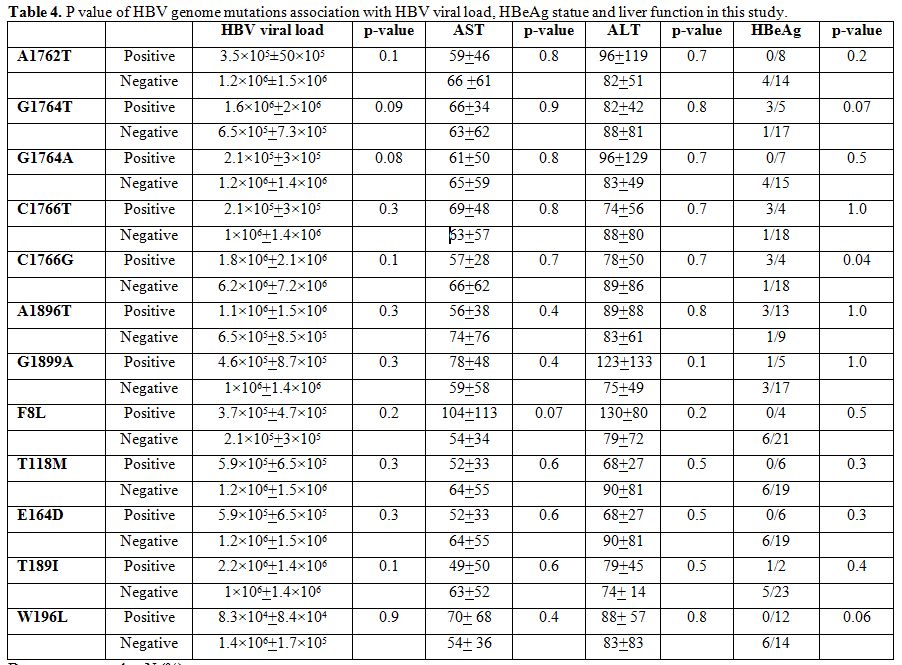 |
Table 4. P value of HBV genome mutations association with HBV viral load, HBeAg statue and liver function in this study. |
 |
Figure 2. UPGMA
phylogenetic tree of surface genes sequences from 33 HBV strains. The
tree rooted with HBV Woolly Monkey (AY226578) sequence. Genetic
distances were estimated using the Kimura 2-parameter matrix.
Clustering of sequences was supported by 1000 resamplings of the data
sets. |
Discussion
Analysis
of the S gene sequence revealed that the majority of isolates (30 out
of 33, 90.9%) belonged to the awy2 subtype while the rest (3 out of 33,
9.1%) were of the awy1 subtype. These findings are in agreement with
other Iranian studies[6,15]
reporting that the awy2 subtype was predominant. However, our results
showed a higher rate of the awy1 subtype than previously reported.[6,15]
Hepatitis
B surface antigen encompasses several B, Th, and CTL epitopes. The
significance of substitution within these immune epitopes in the
pathogenesis of chronic HBV is controversial.[7]
Mutations occurring within the MHR, especially the "a determinant"
domain, may alter the antigenicity; thus, this conformational change
may contribute to false-negative serological tests, the presence of
occult hepatitis, escaping vaccine-induced immunity and failure of the
HBV immunoglobulin (HBIg) therapy.[16-18] Viruses
harboring mutated T-cell epitopes may not be recognized by the T-cell
of an individual; thus, it will not increase anti-HBs production and,
it can result in the progression of chronicity.[9]
The
result of this study indicated that most of the amino acid changes (22
out of 32, 70.0%) appeared in different immune epitopes, of which 6
(27.0%) and 16 (72.0%) were located in the B cell and Th epitopes,
respectively. These results are in line with the finding by Moradi et
al.,[19] showing the most occurrence of mutations
(42.5%) in the Th immune epitope. However, it is in contrast to
observations by Norouzi et al.[20] and Khedive et al.[7] stating that most of the mutations were clustered in the CTL immune epitope.
A
prevalent mutation in the PC region of the HBV genome is a substitution
at position G1896A (codon 28), resulting in the disappearance of HBeAg.[21]
The rate of mutation at residue 1896 correlates with the HBV genotype
and varies geographically. The pre-core mutations are more frequently
seen in the D genotype and are more often observed in the Mediterranean
region.[22]
In our study, G1896A mutation was
detected in 16 (61.0%) out of 26 isolates (12 CHB patients, 1
cirrhosis, 3 inactive carriers). This finding is in line with other
studies in France in 252 HBsAg positive carriers[23] and Korea in 472 patients with chronic HBV infection,[24]
which reported the substitution of G1896A in 54.9% and 55.0% of
subjects, respectively. However, the present study showed a higher rate
of G1896A than other studies in Iran by Ghabeshi et al. in 50 CHB
patients,[25] Moradi et al. in 120 CHB patients[26] and Soleimani in 69 CHB patients[27]
which demonstrated the presence of G1896A in 46.0%, 36.66% and 17.3% of
patients, respectively. It was speculated that this higher frequency
might be affected by the host immune system.
In this study,
another common pre-core mutation at position G1899A was detected in 6
(23.0%) patients (4 CHB patients, 1 cirrhosis, 1 inactive carrier). It
was observed that all subjects with the G1899A variant carried G1896A.
Some studies revealed that G1899A is found to be associated with the
severity of liver diseases.[28,29] Our finding is comparable with a study which showed G1899A in 29.3% of patients.[23]
Another study performed in Korea indicated that all isolates with a G
to A change at position 1899, had a concomitant G1896A change.[24]
A
predominant double mutation in the basic core promoter region involves
a G to A change at nucleotide 1764 and an A to T change at nucleotide
1762. This mutation may cause a decrease in the HBeAg level-up to 70.0%
and increase viral genome replication.[30] The
mechanism by which the G1764A/A1762T dual mutation enhances the
virulence of HBV is not fully understood. It is thought that this
double mutation forms a new binding site for the hepatocyte nuclear
factor 1 (HNF1), leading to a reduced pre-core RNA expression and
enhanced pre-genomic RNA transcription.[31,32]
The
result of the present study revealed the presence of A1762T and G1764A
mutations in 30% (4 CHB patients, 3 cirrhosis, 1 inactive carrier) and
26% (3 CHB patients, 3 cirrhosis, 1 inactive carrier) of subjects,
respectively. The proportion of patients with A1762T/G1764A dual mutant
in our study (23.0%,6/26) (3 CHB patients, 3 cirrhosis) is
consistent with recent findings from Iran (19.6%),[26] Malaysia (26.9%) in 93 HBV carriers(26.9%)[33] and Morocco in 221 chronic carriers(22.9%).[34]
Another
frequent dual mutation detected in the present study was a G to T
change at nucleotide 1764 and a C to G change at nucleotide 1766. It
has been suggested that the G1764T/C1766G mutant creates a new binding
site for the hepatocyte nuclear factor 3 (HNF3), and increases core
promoter activity.[35]
In our data, the
G1764T/C1766G mutant was seen in 26.0% (7/26) of patients (6 CHB
patients, 1 inactive carrier). None of the patients with G1764T/C1766G
mutation carried A1762T/G1764A substitution. This finding is similar to
the study by Sendi et al. in 97 CHB patients with HBeAg negative
reporting that 30.0% of subjects had G1764T/C1766G double mutation, and
the combined mutational patterns T1762/A1764/ G1766 or
T1762/T1764/G1766 which would not generate binding sites for HNF1 or
HNF3 were not seen.[35] Some studies revealed that
the A1762/G1764A mutant accompanied by G1757A is associated with lower
viral load and ALT level; hence, G1757A acts as an inhibitor to the
A1762/G1764A mutant.[31] Nevertheless, the
simultaneous presence of the A1762T/G1764A in conjunction with G at
position 1757 is more efficient. When there is G1757A, the
C1766G/G1764T double mutant is more efficient than the A1762T/G1764A
mutation.[35] More extensive research work is needed to explore the tendency to either A1762T/G1764A or C1766G/G1764T.
Conclusions
Our
results showed that most mutations within the S region were clustered
in the Th immune epitope. Furthermore, the present data indicate a high
rate of G1896A mutant in the PC region among Iranian CHB patients and a
negative correlation between the emergence of A1762T/G1764A mutation
and G1764T/C1766G mutant in the BCP region.
Acknowledgment
This study has been supported by the infectious disease Research Center, Shahid Beheshti University of Medical Sciences.
References
- Agarwal A, Sen S, Banerjee D, Srivastava R,
Praharaj A: Distribution of hepatitis B virus genotype and cancer
predicting precore and basal core promoter mutations. Medical Journal
Armed Forces India 2015, 71(3):225-232. https://doi.org/10.1016/j.mjafi.2015.04.003 PMid:26288490 PMCid:PMC4534539
- Wang
X-L, Ren J-P, Wang X-Q, Wang X-H, Yang S-F, Xiong Y: Mutations in
pre-core and basic core promoter regions of hepatitis B virus in
chronic hepatitis B patients. World Journal of Gastroenterology 2016,
22(11):3268. https://doi.org/10.3748/wjg.v22.i11.3268 PMid:27004005 PMCid:PMC4790003
- Assih
M, Ouattara AK, Diarra B, Yonli AT, Compaore TR, Obiri-Yeboah D, Djigma
FW, Karou S, Simpore JJWjoh: Genetic diversity of hepatitis viruses in
West-African countries from 1996 to 2018. World J Hepatol. 2018 Nov
27;10(11):807-821 https://doi.org/10.4254/wjh.v10.i11.807 PMid:30533182 PMCid:PMC6280160
- Echevarría JM, Avellón A: Hepatitis B virus genetic diversity. Journal of Medical Virology 2006, 78(S1):S36-S42. https://doi.org/10.1002/jmv.20605 PMid:16622876
- Pujol
FH, Navas M-C, Hainaut P, Chemin I: Worldwide genetic diversity of HBV
genotypes and risk of hepatocellular carcinoma. Cancer Letters 2009,
286(1):80-88. https://doi.org/10.1016/j.canlet.2009.07.013 PMid:19683385
- Pourkarim
MR, Sharifi Z, Soleimani A, Amini‐Bavil‐Olyaee S, Elsadek Fakhr A,
Sijmons S, Vercauteren J, Karimi G, Lemey P, Maes P: Evolutionary
analysis of HBV "S" antigen genetic diversity in Iranian blood donors:
a nationwide study. Journal of Medical Vrology 2014, 86(1):144-155. https://doi.org/10.1002/jmv.23798 PMid:24150816
- Khedive
A, Norouzi M, Ramezani F, Karimzadeh H, Alavian S, Malekzadeh R,
Montazeri G, Nejatizadeh A, Ziaee M, Abedi F: Hepatitis B virus surface
protein mutations clustered mainly in CTL immune epitopes in chronic
carriers: results of an Iranian nationwide study. Journal of Viral
Hepatitis 2013, 20(7):494-501. https://doi.org/10.1111/jvh.12045 PMid:23730843
- Petit
M-A, Maillard P, Capel F, Pillot J: Immunochemical structure of the
hepatitis B surface antigen vaccine-II. Analysis of antibody responses
in human sera against the envelope proteins. Molecular Immunology 1986,
23(5):511-523. https://doi.org/10.1016/0161-5890(86)90114-8
- Ramezani
F, Norouzi M, Sarizade GR, Poortahmasebi V, Kalantar E, Magnius L,
Norder H, Domingo E, Jazayeri SM: Mutation hot spots in hepatitis B
surface antigen in chronic carriers from Khoozestan province, southern
of Iran. Iranian Journal of Allergy, Asthma and Immunology 2013,
12(3):269.
- Caligiuri
P, Cerruti R, Icardi G, Bruzzone B: Overview of hepatitis B virus
mutations and their implications in the management of infection. World
Journal of Gastroenterology 2016, 22(1):145. https://doi.org/10.3748/wjg.v22.i1.145 PMid:26755866 PMCid:PMC4698481
- Hunt CM, McGill JM, Allen MI, Condreay LD: Clinical relevance of hepatitis B viral mutations. Hepatology 2000, 31(5):1037-1044. https://doi.org/10.1053/he.2000.6709 PMid:10796877
- Kramvis A, Kew M: The core promoter of hepatitis B virus. Journal of Viral Hepatitis 1999, 6(6):415-427. https://doi.org/10.1046/j.1365-2893.1999.00189.x PMid:10607259
- Bozdayı
AM, Bozkaya H, Türkyılmaz AR, Sarýodlu M, Çetinkaya H, Karayalçın S,
Yurdaydın C, Uzunalimoğlu Ö: Nucleotide divergences in the core
promoter and precore region of genotype D hepatitis B virus in patients
with persistently elevated or normal ALT levels. J Clin Virol. 2001
Apr;21(1):91-101 https://doi.org/10.1016/S1386-6532(01)00148-2
- Jazayeri
M, Basuni A, Sran N, Gish R, Cooksley G, Locarnini S, Carman WF: HBV
core sequence: definition of genotype‐specific variability and
correlation with geographical origin. J Viral Hepat. 2004
Nov;11(6):488-501 https://doi.org/10.1111/j.1365-2893.2004.00534.x PMid:15500549
- Mohebbi
S, Amini‐Bavil‐Olyaee S, Zali N, Noorinayer B, Derakhshan F, Chiani M,
Nejad MR, Antikchi M, Sabahi F, Zali M: Molecular epidemiology of
hepatitis B virus in Iran. Clinical Microbiology and Infection 2008,
14(9):858-866. https://doi.org/10.1111/j.1469-0691.2008.02053.x PMid:18844687
- Ma
Q, Wang Y: Comprehensive analysis of the prevalence of hepatitis B
virus escape mutations in the major hydrophilic region of surface
antigen. Journal of Medical Virology 2012, 84(2):198-206. https://doi.org/10.1002/jmv.23183 PMid:22170538
- Echevarría
JM, Avell0ón A: Improved detection of natural hepatitis B virus surface
antigen (HBsAg) mutants by a new version of the VITROS® HBsAg assay.
Journal of Medical Virology 2008, 80(4):598-602. https://doi.org/10.1002/jmv.21146 PMid:18297712
- Diarra
B, Yonli AT, Sorgho PA, Compaore TR, Ouattara AK, Zongo WA, Tao I,
Traore L, Soubeiga ST, Djigma FWJMjoh et al: Occult hepatitis B virus
infection and associated genotypes among HBsAg-negative subjects in
Burkina Faso. Mediterr J Hematol Infect Dis. 2018 1;10(1):e2018007.
doi: 10.4084/MJHID.2018.007. eCollection 2018. https://doi.org/10.4084/mjhid.2018.007 PMid:29326804 PMCid:PMC5760064
- Moradi
A, Zhand S, Ghaemi A, Javid N, Tabarraei A: Mutations in the S gene
region of hepatitis B virus genotype D in Golestan Province-Iran. Virus
Genes 2012, 44(3):382-387. https://doi.org/10.1007/s11262-012-0715-z PMid:22274739
- Norouzi
M, Ghorashi SA, Ataei B, Yaran M, Malekzadeh R, Alavian SM, Judaki MA,
Ghamari S, Namazi A, Rahimnia R: Hepatitis B virus surface antigen
variants clustered within immune epitopes in chronic hepatitis B
carriers from Hormozgan Province, south of Iran. Iranian Journal of
Basic Medical Sciences 2010, 13(4):213-224.
- Nordin
M, Ingman M, Lindqvist B, Kidd‐Ljunggren K: Variability in the precore
and core promoter region of the hepatitis B virus genome. Journal of
Medical Virology 2014, 86(3):437-445. https://doi.org/10.1002/jmv.23839 PMid:24249691
- Constantinescu
I, Dinu A-A, Boscaiu V, Niculescu M: Hepatitis B Virus Core Promoter
Mutations in Patients With Chronic Hepatitis B and Hepatocellular
Carcinoma in Bucharest, Romania. Hepatitis monthly 2014, 14(10). https://doi.org/10.5812/hepatmon.22072 PMid:25477976 PMCid:PMC4250966
- Ducancelle
A, Pivert A, Bertrais S, Boursier J, Balan V, Veillon P,
Guillou‐Guillemette H, Thibault V, Castelain S, Roquebert B: Different
precore/core mutations of hepatitis B interact with, limit or favor
liver fibrosis severity. Journal of gastroenterology and hepatology
2016. https://doi.org/10.1111/jgh.13338 PMid:26992056
- Yoo
BC, Park J-W, Kim HJ, Lee DH, Cha YJ, Park SM: Precore and core
promoter mutations of hepatitis B virus and hepatitis B e
antigen-negative chronic hepatitis B in Korea. Journal of Hepatology
2003, 38(1):98-103. https://doi.org/10.1016/S0168-8278(02)00349-5
- Ghabeshi
S, Sharifi Z, Hosseini SM, Shooshtari MM: Correlation between viral
load of HBV in chronic hepatitis B patients and precore and Basal core
promoter mutations. Hepatitis Monthly 2013, 13(2). https://doi.org/10.5812/hepatmon.7415 PMid:23599717 PMCid:PMC3628088
- Moradi
A, Zhand S, Ghaemi A, Javid N, Bazouri M, Tabarraei A: Mutations in
pre-core and basal-core promoter regions of hepatitis B virus in
chronic HBV patients from Golestan, Iran. Iranian Journal of Basic
Medical Sciences 2014, 17(5):370.
- Fariba
Soleimani, Seyed Ali Mohammmad Arabzadeh, Hamidreza Mollaie, ZahraI
ranmanesh, Najmeh Nikpour, Motahar M: Evaluation of the frequency of
precore/core mutation in patients with chronic hepatitis B, Kerman,
Southeast of Iran. Asian Pacific Journal of Tropical Disease 2016,
6(8):603-607. https://doi.org/10.1016/S2222-1808(16)61093-9
- Tillmann
H, Trautwein C, Walker D, Michitaka K, Kubicka S, Böker K, Manns M:
Clinical relevance of mutations in the precore genome of the hepatitis
B virus. Gut 1995, 37(4):568-573. https://doi.org/10.1136/gut.37.4.568 PMid:7489947 PMCid:PMC1382912
- Chan
HL, Leung NW, Hussain M, Wong ML, Lok AS: Hepatitis B e
antigen-negative chronic hepatitis B in Hong Kong. Hepatology 2000,
31(3):763-768. https://doi.org/10.1002/hep.510310330 PMid:10706570
- Fang
ZL, Ling R, Wang SS, Nong J, Huang CS, Harrison TJ: HBV core promoter
mutations prevail in patients with hepatocellular carcinoma from
Guangxi, China. Journal of Medical Virology 1998, 56(1):18-24. https://doi.org/10.1002/(SICI)1096-9071(199809)56:1<18::AID-JMV4>3.0.CO;2-Q
- Poustchi
H, Mohamadkhani A, Bowden S, Montazeri G, Ayres A, Revill P, Farrell G,
Locarnini S, George J, Malekzadeh R: Clinical significance of precore
and core promoter mutations in genotype D hepatitis B‐related chronic
liver disease. Journal of Viral Hepatitis 2008, 15(10):753-760. https://doi.org/10.1111/j.1365-2893.2008.00998.x PMid:18507754
- Fujiwara
K, Tanaka Y, Orito E, Ohno T, Kato T, Sugihara K, Hasegawa I, Sakurai
M, Ito K, Ozasa A: Distribution of HBV genotypes among HBV carriers in
Benin: phylogenetic analysis and virological characteristics of HBV
genotype E. World Journal of Gastroenterology: WJG 2005,
11(41):6410-6415. https://doi.org/10.3748/wjg.v11.i41.6410 PMid:16425408 PMCid:PMC4355778
- Suppiah
J, Zain RM, Bahari N, Nawi SH, Saat Z: G1896A Precore Mutation and
Association With HBeAg Status, Genotype and Clinical Status in Patients
With Chronic Hepatitis B. Hepatitis Monthly 2015, 15(10). https://doi.org/10.5812/hepatmon.31490 PMid:26587040 PMCid:PMC4644636
- Baha
W, Ennaji MM, Lazar F, Melloul M, El Fahime E, El Malki A, Bennani A:
HBV genotypes prevalence, precore and basal core mutants in Morocco.
Infection, Genetics and Evolution 2012, 12(6):1157-1162. https://doi.org/10.1016/j.meegid.2012.04.026 PMid:22579480
- Sendi
H, Mehrab-Mohseni M, Zali MR, Norder H, Magnius LO: T1764G1766 core
promoter double mutants are restricted to Hepatitis B virus strains
with an A1757 and are common in genotype D. Journal of General Virology
2005, 86(9):2451-2458. https://doi.org/10.1099/vir.0.81023-0 PMid:16099903
[TOP]