Irene Motta1,2, Valentina Ghiaccio3, Andrea Cosentino2 and Laura Breda3.
1 Department of Clinical Sciences and Community Health, Università degli Studi di Milano, Milan, Italy.
2 Fondazione IRCCS Ca' Granda, Ospedale Maggiore Policlinico, Milan, Italy.
3 Department of Pediatrics, Division of Hematology, Children's Hospital of Philadelphia, Philadelphia, PA, United States.
Correspondence to: Laura Breda. Department of Pediatrics, Division of
Hematology, Children's Hospital of Philadelphia, Philadelphia, PA,
United States. E-mail:
bredal@email.chop.edu
Published: November 1, 2019
Received: August 29, 2019
Accepted: October 22, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019067 DOI
10.4084/MJHID.2019.067
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Inherited
hemoglobin disorders, including beta-thalassemia (BT) and sickle-cell
disease (SCD), are the most common monogenic diseases worldwide, with a
global carrier frequency of over 5%.[1] With
migration, they are becoming more common worldwide, making their
management and care an increasing concern for health care systems.
BT
is characterized by an imbalance in the α/β-globin chain ratio,
ineffective erythropoiesis, chronic hemolytic anemia, and compensatory
hemopoietic expansion.[1] Globally, there are over
25,000 births each year with transfusion-dependent thalassemia (TDT).
The currently available treatment for TDT is lifelong transfusions and
iron chelation therapy or allogenic bone marrow transplantation as a
curative option. SCD affects 300 million people worldwide[2]
and severely impacts the quality of life of patients who experience
unpredictable, recurrent acute and chronic severe pain, stroke,
infections, pulmonary disease, kidney disease, retinopathy, and other
complications. While survival has been dramatically extended, quality
of life is markedly reduced by disease- and treatment-associated
morbidity.
The development of safe, tissue-specific and
efficient vectors, and efficient gene-editing technologies has led to
the development of several gene therapy trials for BT and SCD. However,
the complexity of the approach presents its hurdles. Fundamental
factors at play include the requirement for myeloablation on a patient
with benign disease, the age of the patient, and the consequent bone
marrow microenvironment. A successful path from proof-of-concept
studies to commercialization must render gene therapy a sustainable and
accessible approach for a large number of patients. Furthermore, the
cost of these therapies is a considerable challenge for the health care
system. While new promising therapeutic options are emerging,[3,4] and many others are on the pipeline,[5]
gene therapy can potentially cure patients. We herein provide an
overview of the most recent, likely potentially curative therapies for
hemoglobinopathies and a summary of the challenges that these
approaches entail.
|
Bone Marrow Transplantation (BMT)
One
of the most recurring statements in the literature about hematopoietic
stem cell transplantation (HSCT) in thalassemia is “Allogenic bone
marrow transplantation is the only available curative treatment for
thalassemia major”. This approach is rooted in the Italian experience
of the early 1980s at the transplant center of Pesaro, where throughout
the 1980s and early 1990s, more than 1000 thalassemia major (TM)
patients received transplants, and the center reported a 20-year
probability of Thalassemia-free survival of 73% in 900 consecutive
transplanted patients.[6]
More than 35 years have
passed since the first TM patient underwent an allogeneic BM
transplantation. Since the pioneering Pesaro experience, the curative
role of BM transplantation in TM has been well established, and other
European Bone Marrow Transplant (EBMT) centers have routinely performed
this therapy.
Indeed, in a retrospective non-interventional
study, data analyzed from the EBMT registry database on 1493
consecutive patients with TM transplanted between 1 January 2000 and 31
December 2010. The 2-year overall survival (OS) and event-free survival
(EFS) were 88±1% and 81±1%, respectively, after a median observation
period of 2 years. OS and EFS were 90%, 81% and 93% (P<0.001), and
82%, 76%, and 85% (P=0.003) in patients who had received bone marrow,
peripheral blood, or cord blood (alone or combined), respectively.[7]
The
decision to undergo a curative but potentially fatal treatment should
be taken with an assessment of transplantation-related mortality (TRM).
The Pesaro risk assessment (for a pediatric population) was developed
in the early 1990s.[8] This assessment stratifies the
outcome of transplantation in three classes on the basis of
hepatomegaly, portal fibrosis, and irregular chelation history.[6]
According to these risk factors, patients were categorized into three
risk classes: Class 1 patients, who had none of these adverse risk
factors, class 2 patients, who had one or two adverse risk factors and
class 3 patients, who had all three. The thalassemia-free survival
(TFS) was respectively 85-90%, 80% and 65-70%, while the
transplant-related mortality (TRM) positively increased between classes
1, 2, and 3.[9]
Taking these results into
consideration, it is mandatory to offer an HSCT to a young TM patient
with a matched sibling donor before the development of iron overload
and iron tissue damage. Of note, all the gene therapy clinical trials
have as exclusion criteria the presence of a matched sibling donor
(MSD).
The rational of bone marrow transplantation in a TM patient
is to restore the tissue's capability of producing functional
hemoglobin, and that can be achieved even with the coexistence of donor
and recipient hematopoietic stem cells. In fact, in approximately 10%
of patients, a condition of persistent mixed chimerism in which the
donor hematopoiesis maintain the potential to correct the phenotype of
the disease was described. On the other hand, it is vital to minimize
the risk of graft-versus-host disease (GVHD), since the immunologic
effect of the transplant is unnecessary in a non-malignant disease such
as thalassemia.
According to the retrospective EBMT survey
concerning the HLA matching of transplant recipients, 1061 (71.1%) HSCT
were performed using HLA-identical sibling donors, 127 (8.5%) from
another HLA-matched relative, 57 (3.8%) from an unmatched relative and
210 (14.1%) from a matched unrelated donor.[7] This
significant disproportion of donor sources is due to the fact that MSD
transplants give the safest outcome with a 2-year OS of 91%, compared
to 88% of matched family donors and 77% of matched unrelated donors.
In
a recent survey, Li et al. analyzed data about BMT in children and
young adults with TM comparing the use of alternative donors and
HLA-matched related donor in 3 geographic regions: China, India, and
the United States. They reported that the 5-year probabilities of OS
after HLA-matched relative, HLA-mismatched relative, HLA-matched
unrelated, and HLA-mismatched unrelated donor transplants were 89%,
73%, 87% and 83%, respectively. The 5-year probabilities of EFS after
HLA-matched relative, HLA-mismatched relative, HLA-matched unrelated,
and HLA-mismatched unrelated donor transplants were 86%, 70% and 82%,
78%, respectively.[10] This report is the first
demonstrating comparable event-free and overall survival after
HLA-matched related and HLA-matched unrelated donor transplantation.
BM
transplantation was the only approved curative treatment for TM patient
until June 2019, when the first gene therapy product, Zynteglo, was
approved by the European Medicine Agency (EMA) for TDT patients who do
not entirely lack beta-globin and who are eligible for stem cell
transplantation but do not have a matching related donor.[11]
Of note, the availability of a suitable donor, together with patient
fitness, are the main limitations to the broader use of HSCT. For this
reason, there is a need to reduce the toxicity of HSCT, investigate new
conditioning strategies, or improve and extend the autologous gene
therapy approach also to the most severe cases..
Clinical Trials Based on Gene Therapy and Gene Editing
Currently,
a growing number of clinical trials for hemoglobinopathies are
investigating the safety and efficacy of gene addition and gene editing
based technologies to rescue hemoglobin synthesis in
beta-globinopathies. The first and longer-dated include trials
indicated for the cure of beta-thalassemia as well as sickle cell
disease via beta or gamma-globin gene addition, while the latter is
mostly aimed at the cure of SCD via reactivation of fetal hemoglobin.
The first gene addition-based trial that led to cure in a beta0/betaE
patient was reported in 2010,[12] and since then,
roughly 50 patients with BT and more than 20 patients with SCD have
been treated, and the related outcomes have been reported. The data
published or communicated on the trials were obtained with four
different lentiviral vectors: HPV569, later implemented as Lentiglobin
BB305,[13] employed by bluebird bio (formerly Genetix Pharmaceuticals); GLOBE,[14,15] employed by IRCCS San Raffaele, Italy; RVT-1801, employed by CCHMC, Cincinnati, USA (presented at the 22nd ASCGT meeting in 2019), and TNS9.3.55,[16]
employed by Memorial Sloan Kettering Cancer Center NY, USA. This trial
(NCT01639690) was subsequently halted, and only one patient (out of 4
treated) had a significant decrease in transfusion requirements. An
additional trial has been initiated by UCLA, using the AS3-FB vector,[17] but no data related to patients’ outcome has been released so far. Table 1
lists the trial identifiers as well as the specifics of vectors and
regimens employed in each trial. The significant challenges for these
trials relate to two major determinants intrinsic to
hemoglobinopathies: the severity of the hemoglobin defect in BT (i.e.,
beta0/0 genotype being the most challenging to correct) as well as the
disease progression for both BT and SCD. Initial trials, focused on
adult patients with a beta+/+ or beta+/0 BT, had a fair degree of
success, where patients became mostly transfusion independent even with
suboptimal level (less than two copies per genome) of lentiviral
integration or vector copy number (VCN) (NCT01745120 and NCT02151526).
This level of integration proved to be unsuccessful at achieving
transfusion independence for most of the adult patients affected by BT
due to a beta0/0 genotype.[18] Therefore, subsequent
trials have aimed at optimizing transduction protocols to obtain higher
VCN (roughly 3-4 copies) in the drug product (DP), which indicates a
patient hematopoietic stem cell (HSC) transduced with the lentiglobin
vector, and maximizing transgenic chimerism, which indicates the
proportion of HSC successfully transduced and engrafted. Patients with
beta0/0 BT and SCD have benefited from the new transduction regimens,
achieving hemoglobin (Hb) levels that do not require transfusion and
therefore are considered curative (NCT02906202 and groups B and C of
NTC02140554, see Table 1 for
details). In light of these results, EMA recently approved bluebird
bio’s Zintheglo, the first drug for gene therapy in TDT, in Europe. We
learned that disease progression, hence the age of a patient, can
greatly affect the outcome of gene therapy. Actually, the trial
conducted in Italy (NCT02453477), using GLOBE, showed that the same
protocol produced the best outcome in the youngest cohort (pediatric),
while it was progressively less successful for the middle and oldest
ones.[19] More data on the pediatric population will be necessary to better elucidate the correlation between curative outcome and age.
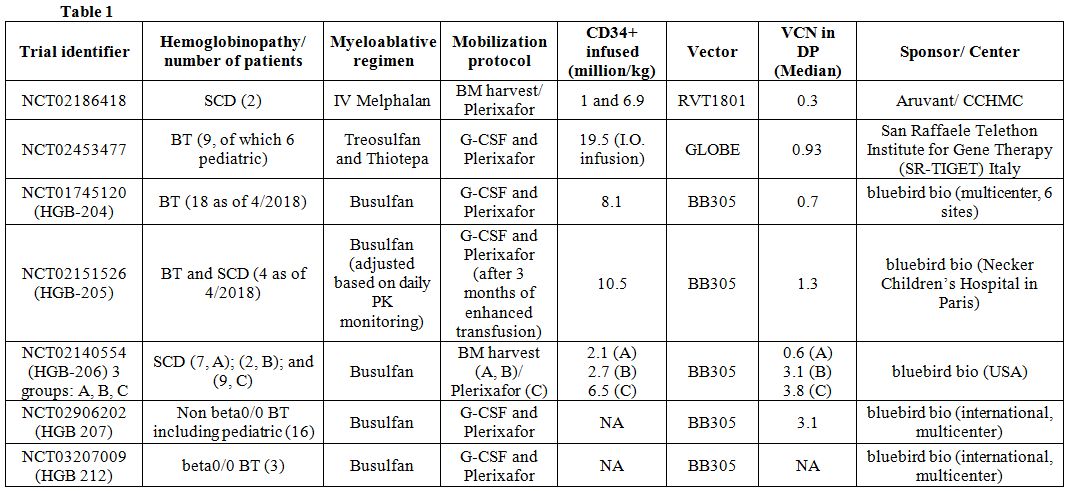 |
Table 1 |
Additionally,
in the first seven subjects with SCD (NTC02140554-group A), for whom
BM-derived HSC were utilized, the median expression of vector-derived
hemoglobin was only 0.4 g/dL (range 0.1 to 1 g/dL). Both cases suggest
that the BM environment in hemoglobinopathies is not fully understood,
and its physiology might be progressively altered by disease
progression. Better results were achieved by the procurement of HSC
from peripheral blood via apheresis, using improved mobilization
practices, as discussed in the section that follows.
The first
clinical data based on a gene-editing approach were recently
communicated for the clinical trial NCT03282656 for severe SCD (ASH
2018), which has been conducted at the Boston Children’s Hospital, MA,
by bluebird bio. As for most of the gene addition studies above
mentioned, this pilot trial uses Plerixafor to mobilize patients’ HSC
and a busulfan regimen for conditioning. The first three patients
presented at ASH 2018 were infused with a drug product (HSC count: from
3.3 to 6.7 X 10^6/kg weight) that had a VCN between 3.25 and 5. The
drug product was transduced (over 90% rate) with BCH_BB-LCRshRNA (miR),[13] a lentiviral vector that carries a short hairpin RNA targeting BCL11A, a known repressor of gamma-globin expression.[20]
The first patient treated and followed for six months presented a 24%
increase in HbF, a high number of F expressing cells in circulation
(72%), a considerable reduction in reticulocytosis as well early
mitigation of some of the cellular phenotype of SCD. Enrollment for
this trial is still active and will involve a total of 7 patients.
Since
naturally-occurring deletional mutations of the binding site of BCL11A
in the gamma-globin promoter have been found to be associated with
cases of high persistence of fetal hemoglobin (HPFH), this very same
deletion has been the target of the newest gene-editing approaches for
SCD. Two trials, enrolling patients in more than one site within the
US, NCT03745287 and NCT03653247, are been conducted by CRISPR
Therapeutics and Bioverativ (Sanofi), respectively.
Although
based on the same target, the CRISPR therapeutics approach relies on
the use of CRISPR/CAS9 to edit patients’ HSC, while Sanofi’s approach
relies on the use of a zinc-finger nuclease. An additional trial by
Sangamo, NCT03432364, also uses a zinc-finger nuclease approach to
target the enhancer of BCL11A, although this treatment is indicated for
BT. Preliminary results, presented by Sangamo at ASCGT 2019, showed
successful gene editing of peripheral white blood cells collected from
the first treated patient, a beta0/0 BT case, an increase of fetal Hb
levels, and stable total Hb values around 9g/dL at 50 days from the
infusion. Longer follow-up periods for all these most recent studies
are needed to further elucidate the extent of the success of the
gene-editing technology for hemoglobinopathies.
Conditioning in Clinical Trials and New Preclinical Development
The
conditioning regimen for hemoglobinopathies does not include drugs with
specific antitumor effects, given the non-malignant nature of these
diseases.[21] Nevertheless, to achieve stable
engraftment, patients receive chemotherapy that aims to make space,
eliminating an extremely proliferating marrow with erythroid
hyperplasia and favor engraftment of HSC.[22,23]
Data
from reduced-intensity allogeneic transplantation in limited clinical
series show that the achievement of stable donor engraftment is
difficult in immunocompetent patients with hemoglobinopathies.[24,25]
Therefore, patients with BT and SCD need more intensive myeloablative
conditioning regimens in order to reduce the risk of graft failure.[23]
Myeloablative conditioning is associated with short- and long-term
toxicity in hematopoietic and non-hematopoietic tissues, including
cancer and infertility and gonadal failure, particularly among females.[26] The ideal regimen should combine an efficient depletion of HSC and minimized toxicity.
Non-myeloablative
reduced-intensity conditioning, based on busulfan at a total dose of 8
mg/kg, has been used in the gene addition trial (NCT01639690) at
Memorial Sloan Kettering Cancer Center[27] and resulted in insufficient engraftment of gene-marked cells and minimal clinical benefit.[28]
In two phase 1/2 GT trials (NCT01745120 and NCT02151526)
for TDT with LentiGlobin BB305 vector, myeloablative conditioning with
busulfan as a single agent was used. A dose adjustment was performed to
achieve appropriately targeted drug exposure. The average daily plasma
busulfan area-under-the-curve values ranged from 3029 to 4714 μM per
minute in HGB-204 (estimated values) and from 4670 to 5212 μM per
minute in HGB-205 (actual values). Among the 22 treated patients, 9
serious adverse events (SAE) were reported, including two episodes of
veno-occlusive liver disease attributed to busulfan conditioning.[18] Busulfan was also used for SCD patients in several trials (NCT02151526, NCT03282656, NCT02247843) with mixed results.[29]
The
phase 1/2 GT trial for TDT at IRCCS San Raffaele in Milan, used
myeloablative conditioning with treosulfan–thiotepa, given their
reduced extramedullary toxicity compared to busulfan.
Chemotherapy-related toxicity was mild, and 5 SAE of infectious nature
were reported and resolved. Neither veno-occlusive disease nor hepatic
toxicity was observed.[19] Most recently, preliminary
results from a reduced-intensity conditioning (RIC) phase 1/2 pilot
study on gene transfer for SCD were presented.[30]
For this trial, a single dose of melphalan is used as the conditioning
regimen, while patients’ HSC are corrected using a γ-globin based
lentiviral vector (RVT1801; NCT02186418). Results from this trial
showed excellent safety, feasibility, with minimal post-transplant
toxicity, rapid count recovery, and sustained stable genetically
modified cells in peripheral blood and bone marrow.
Although
autologous HSCT using genetically corrected cells would avoid the risk
of graft-versus-host disease (GVHD), the genotoxicity of conditioning
remains a substantial barrier to the development of this approach in
non-malignant disorders. The toxicities of conditioning lessen the
willingness of patients and healthcare providers to consider this
therapy. Novel strategies that aim to reduce regimen-related toxicity,
remaining on the other hand sufficiently myeloablative, are based on
the use of antibodies that specifically target HSC and other
hematopoietic cells in the bone marrow niche, sparing non-hematopoietic
cells.[31,32] If proof of safety and efficacy stands,
these new regimens will satisfy the need for reduction of morbidity and
mortality related to the current conditioning regimens, mainly based on
busulfan administration.
In mice, immunotoxin CD45–saporin (SAP)
allows high-level engraftment and multi-lineage repopulation of
transplanted HSC without the need for chemotherapy or irradiation and
enables efficient engraftment of donor cells and full correction of a
sickle-cell anemia model.[31]
The depletion of
HSC can also be achieved using a c-kit (CD117) antibody, as its
receptor is highly expressed on these cells. Binding of CD117 ligand is
essential for hematopoiesis and self-renewal. The internalization of
the receptor by antibody binding causes HSC failure. A CD117 antibody
molecule conjugated with a non-genotoxic small toxin molecule can
deplete more than 98% of human HSC in NSG mice, leaving intact T and B
cells.[33] Instead, the unconjugated antibody
presents a much-reduced effect. The velocity of the internalization of
the receptor and selectivity of the CD117 molecule makes this method an
excellent candidate for gene-corrected BMT for hematopoietic
conditions, like BT and SCD, preventing the systemic toxicity typical
of irradiation and chemotherapy regimens.
A clinical study,
carried out at Stanford (NCT02963064), utilizes this very strategy to
condition patients with severe combined immunodeficiency disease prior
to HSCT. This study aims at demonstrating the safety and efficacy of
AMG191, a CD117 conjugated antibody given intravenously in one dose,
followed by infusion of donor CD34+CD90+ HSC. This purified fraction of
HSC that can be isolated by flow cytometry has been shown to possess
the stem-like quality necessary to repopulate the BM in long term
studies in nonhuman primates.[34] Patients that
undergo this procedure receive 1X106 CD34+CD90+ cells/kg, a much lesser
dose than that one used when the CD34 marker alone is selected. The
antibody clearance can be monitored via PK study to establish the best
time for the infusion of donor cells without compromising their chance
to engraft the BM niche. This protocol includes subjects with a poor
graft from previous HSCT, indicating that this could be performed in
patients whose prior BMT has not succeeded with chemotherapy agents. If
successfully and safely applied to patients with hemoglobinopathies,
this technology could positively impact not only patients’ health, by
eliminating the myeloablation related toxicity, but also limit the cost
of gene therapy, by limiting the number of cells that needs to be
corrected.
CD34+ Hematopoietic Progenitor Cells Mobilization
Peripheral
stem cells are commonly used for transplantation and have replaced bone
marrow as a source of HSC in most transplants, especially in the
autologous setting.[35] Granulocyte-colony
stimulating factor (G-CSF) is the standard agent to mobilize
hematopoietic stem and progenitor cells (HSPC) for transplantation.
However, peculiar features of hemoglobinopathies, namely splenomegaly
and thrombophilia state, may represent risk factors for adverse events.
G-CSF-related enlargements of the spleen, rarely resulting in splenic
rupture,[36-38] and thrombotic events[39] have been reported.
Moreover,
in adult thalassemia patients iron-overload and consequent oxidative
stress, the suppressive effect of long-term transfusions and chelation
on the stem cell compartment, and the "aged" stem cells, could
compromise the safety and success of HSC procurement.[40] Also, in SCD, the use of G-CSF has been associated with severe and life-threatening vaso-occlusive complications.[41,42]
G-CSF induces an increase in white blood cells count, neutrophils,
endothelial cells, platelets, and coagulation activation, all
mechanisms that contribute to the vaso-occlusive crisis.
Among the
other compounds available, Plerixafor represents a good alternative.
Plerixafor is a bicyclam molecule that selectively and reversibly
prevents the binding of stromal-derived factor-1 (SDF-1) to chemokine
CXC-receptor-4 (CXCR4) on HSPC, inducing their mobilization.[43] Plerixafor has proved to be safe and effective in mobilizing HSC in thalassemia patients.[18,19,44]
It
has been shown that Plerixafor and G-CSF mobilize different primitive
HSC populations, either in thalassemia patients with thalassemia or
healthy donors. Plerixafor-mobilized cells have a “stemness” signature
compared to G-CSF mobilized cells, and the combined use of the two
agents attenuates this “stemness". Furthermore, Plerixafor-mobilized
HSC possess the highest ability to home to hematopoietic niches and
engraft in immunodeficient mice, and their global gene expression
signature highlights their superior in vivo reconstitution activity.[45]
Currently,
several phase 1 and 2 trials are evaluating the safety and efficacy in
collecting a sufficient number of HSC with Plerixafor in SCD patients
(NCT02989701, NCT03226691, NCT02193191, NCT02212535, NCT02140554).
Lagresle-Peyrou’s group published the results of a French trial; no
adverse events were observed administrating Plerixafor in a single-dose
of 240 mcg/kg in three patients who had discontinued hydroxyurea (HU).
Moreover, with single apheresis, they were able to collect a high
number of HSC.[46] Interim results from a Memorial
Sloan Kettering Cancer Center trial with Plerixafor at escalating dose
reported data on 15 patients. Ten were on HU and one on chronic
transfusion regimen. Two serious adverse events (pain crisis) have been
observed at 80 and 240 mcg/kg of Plerixafor, and only 33-50% of
patients, according to different doses, reached the target yield of
HSC.[47] Most recent data on group C from the HGB-206
study (NCT02140554) show that mobilization was effective in SCD
patients with Plerixafor at the dose of 240 mcg/kg. No life-threatening
VOCs after Plerixafor mobilization have been reported.[48]
Three
main considerations can be drawn from the studies on conditioning
conducted thus far. One relates to HU administration prior to the
mobilization. HU reduces the amount of circulating CD34+,[49] is associated with myelosuppression, and did not show any beneficial effect in thalassemia patients.[40,50]
In the French trial, patients discontinued HU 3 months before the
mobilization. However, in the New York trial, no association was
observed between HU and the peak of HSC. The second concerns the
maintenance of HbS levels>30% in order to prevent the vaso-occlusive
crisis. In the French trial (NTC02212535), during the three months
before the mobilization, patients underwent a transfusion or
erythro-exchange program.[46] The third is the timing
of apheresis. The peak of circulating HSC in SCD patients have been
observed at 3-6 hours, earlier compared to healthy donors (6-12 hours)[51] in whom apheresis is recommended to start at 11 hours after Plerixafor administration.
From Clinical Trials to Drug Commercialization, the Challenges of Pivotal GT Studies
Because
of their monogenetic etiology, both BT and SCD are attractive targets
for curative approaches as gene addition and gene editing.
Gene
addition strategies have significantly improved over the past ten years
and have provided the most successful results thus far. Although these
approaches may seem straightforward given the single gene defect and
defined cell target, there are still several hurdles that can impact
their success, as previously reported.[52]
One
of the most relevant challenges is to guarantee a level of functional
beta-globin protein expression that can rescue the complete lack of
endogenous adult hemoglobin protein, like that seen in patients with
beta0/0 BT. The constructs employed in clinical trials utilize large
genomic regulatory elements that are essential to express high and
tissue-specific expression of the gene of interest, and they are
engineered to maximize their safety. The need for such large constructs
on the other side can affect the viral assembly and entry, impacting
the yield of viral particles that can be functionally assembled during
manufacturing, and limiting the number of particles that can
effectively enter the target HSC, respectively.
Viral
vector-based gene therapy has become commonplace in both the laboratory
and the clinic, and it is rapidly evolving towards becoming a curative
treatment option. Currently, several products are under clinical
development, and the European Medicine Agency recently approved the
first lentiviral product (Zynteglo,
based on BB305) for gene addition based therapy for BT. The transition
from clinical development to commercialization adds another layer of
complexity to the picture. Thus, the most pressing concern becomes no
longer to make safe, functional lentiviral vectors solely, but to also
refine current processes in order to achieve both yield and quality
while reducing costs, processing times, and risks to support research,
development, and commercialization.
lentivirus-based gene and
cell therapy products were estimated in 2017 to cost in the range of
U.S. $500,000–1,000,000 per patient,[53] a third of which pertains to the lentivirus manufacturing process itself.
Access
to gene therapy to a large number of patients with a hemoglobinopathy
is highly dependent on a reliable high-scale lentiviral production. The
implementation of scalable vector production protocols is indispensable
to fulfill not only the demand of academic and hospital institutions,
but also of the industry, which is incentivized to satisfy the upsurge
of requests for safe and efficient products.
Lentiviral Vectors Manufacturing
Lentiviral
vector manufacturing technologies are based primarily on fully
transient, adherent processes. These 2-dimension methods are tedious,
time-consuming, and difficult to scale up, with carry-associated issues
of process handling, results inconsistency, and high costs. Small-scale
productions for R&D purposes are performed transfecting adherent
293T cells grown in plasticware (Petri dishes, T-flasks, Cell
Factories, Cell Stacks, or HYPERFlask) in the presence of either PEI or
Calcium Phosphate (CaPho).[54,55]Conversely,
large-scale vector manufacturing generally requires a direct scale-up
from small-scale systems to increase volumes and titers, and reduce
variability. This can be accomplished by using alternative culture
devices (e.g., roller bottles, multi tray cell factories, or
microcarriers in stirred tanks) that provide extended anchorage surface
for adherent 293T cells. Valkama AJ et al.[56]
demonstrated how parameters, such as cell density, pH levels, and
transfection methods, play a delicate role for large-scale lentiviral
production. In their study, they use PALL iCELLis, a compact fixed-bed
bioreactor with an integrated perfusion system, comparing yields
achieved with either CaPho precipitation or PEI transfection methods,
within a range of pH and under glucose and lactate concentration
monitoring. In both transfection methods, they confirmed the critical
role of cell density and pH in the efficiency of productivity,[57]
showing higher production at high cell-density and mildly acidic pH,
with vp cm–2 (vp= viral particle) between 1.31E+08 and 4.85E+08 using
iCELLis Nano fixed-bed bioreactors.To
overcome the need for traditional large-scale technologies in
lentiviral production, some investigators have engineered novel
suspension-adapted cell lines.[58] Although there are
no established and standardized methods based on this suspension
approach with a Good Manufacturing Practices (GMP) grade approval,
several studies are ongoing, aiming to fill the demand for a robust
supply chain for GMP materials. Suspension-adapted
cell lines can easily grow at high cell densities in a packed-bed
bioreactor, stirred tanks, or Erlenmeyer flasks agitated on orbital
shakers, which require minimum handling or supplementary laboratory
equipment.[55] Several cell lines (293T, 293FT, and
293SF-3F6) have already been used for the purpose, through a
modification that allows growth in suspension in chemically defined
media (Freestyle 293 and F17, Invitrogen; HyQSFM4TransFx293).[58,59]
Suspension-adapted cell lines grow promptly rendering, thus their
maintenance and propagation much easier than that of adherently growing
cells. Another advantage of the use of this system in clinical
manufacturing is the lack of requirement for bovine serum and animal
origin components in the culture media, which decreases the risk of
contamination by adventitious agents.The
preferential vessel for large-scale lentiviral production using
suspension-adapted cells is a bioreactor. In this type of system, DNA
precipitation using calcium phosphate is expected to be less effective
for transfecting suspension cells because of continuous culture
stirring. Other transfection agents like cationic polymers are used
instead. Using linear or branched 25-kDa PEI, Durocher et al. achieved
the highest transfection efficiency in 293T and 293-EBNA1 cells,
leading to 75% of transfected cells using a green fluorescent protein
(GFP) reporter plasmid.[60] McCarron et colleagues were able to reach yields of up to 109
TU/mL (TU= transducing units) using a packed-bed bioreactor system,
suggesting a more significant potential on the basis that the total
packed-bed surface area is 18m2, which is equivalent to approximately 28 standard 10-layer cell factories.[61]The
viral supernatant is collected from suspension-adapted cells in the
bioreactor by anion exchange chromatography. The eluted material can
then be concentrated using tangential flow filtration (TFF), and the
vector may then be diafiltered into its final formulation buffer.[62]More
recently, developed protocols for lentiviral manufacturing employ
hollow fiber bioreactor, suspension culture processes, and the
implementation of stable producer cell lines. The use of stable
producer cell lines allows to further scale-up production, having the
advantage to remove any undesirable process-derived contaminant, such
as plasmid or host cell DNA or host cell proteins.[63]
The biomedical industry is the driving force to make the use of all
these technologies accessible for large-scale platforms in terms of
manufacturing, costs, and GMP-compliance. Some of these innovative
technologies were presented last May at the 22nd
ASCGT meeting. Two independent companies showed the generation of a
suspension-adapted cell line GMP-compliant for lentiviral packaging
using an inducible Tet system,[64] and production of
lentiviruses up to 200L in PALL iCELLis500, reaching 80.1% transduction
efficiency in T-cells using a Multiplicity of Infection MOI=4,[65] respectively.
HCS Transduction and Handling
Downstream the lentiviral production per se,
the manufacturing of the drug product (DP), which consists of patients’
HSC corrected with a lentivirus, represents another benchmark for
innovative and unconventional protocols that aim to make the process
more efficient and scalable. Efficient HSC transduction requires highly
concentrated and purified lentiviral particles and, in diseases like
beta0/0 BT, in which a higher number of integrations are necessary to
achieve curative outcomes, integration of large transgenic cassette can
be particularly challenging. Resistance to infection has been
attributed to the quiescent (G0) phase and the innate immune defenses
against viral transduction.[66] Given the need to
achieve optimal integration in the DP that guarantees curative effects
without compromising genome safety, several experimental transduction
protocols, and agents to enhance/modulate lentiviral gene transfer
yield have been developed. The necessity to make the manufacturing of
the DP as consistent and reproducible as possible has been driving
force for these technologies.Transduction
efficiency can be improved, even at low MOIs, using small molecules
like cationic-liposomes, polycations, or small peptides as
adjuvant-enhancer of transduction. For over a decade, cationic polymers
such as hexadimethrine bromide (Polybrene) have been employed in
laboratory-based protocols to neutralize the nonspecific electrostatic
interactions between virus and target cell.[67,68]
Due to its negative effect on cell growth and viability, polybrene is
not indicated for clinical use. Therefore, new molecules have been
engineered to promote vector-cell contacts similarly. Balancing safety
and transduction efficiency has shown to be pivotal in the process of
providing new adjuvants. More recently, Vectofusin-1,[69] protamine sulfate,[70] and fibronectin fragments[71]
have shown to increase transduction efficiency by facilitating adhesion
and fusion between the viral cap side and the cell membrane.LentiBOOSTTM
reagent, a large nonionic amphiphilic molecule (poloxamer), has been
extensively used as a transduction enhancer, with greater activity and
better safety profile than similar known nonionic molecules.[72] Human peripheral blood-derived CD34+ HSC transduced with standard MOI of 10, and LentiBOOSTTM show increased gene expression and VCN proportional to adjuvant concentration with unaltered viability. The use of LentiBOOSTTM is compatible with spinoculation protocols, and its use in combination with protamine sulfate shows an addictive effect.[73] Moreover, the use of RetroNectin and LentiBOOSTTM in human HSC transduction concurrently decrease electrostatic interactions and promote either adhesion/fusion and integration.[74]Prostaglandin E2
(PGE2) is a molecule that enhances transduction prior to nuclear entry
and integration, possibly by interfering in the endocytosis-dependent
pathways.[75] While initially intended as an
anti-apoptotic agent in experiments on HSC, prestimulation with PGE2
surprisingly showed a ~1.5 fold increase of VCN, both in vivo after xenotransplantation.[76]
Additionally, the combination of PGE2 and poloxamer synperonic F108 had
a synergistic effect, increasing gene transfer in primitive HSC by ~10
fold in CD34+ HSPC with a globin-based lentivirus, which is highly
desirable when high VCN is needed. Viral
integration can also be facilitated by molecules that modulate
intracellular processes, like Rapamacyn (Rapa). By inducing autophagy
through allosteric inhibition, Rapa enhances post-binding endocytic
events, increasing lentiviral entry, reverse transcription, and genomic
integration.[77] In mouse and human HSC, transduction
with 5 to 20 μg/mL of Rapa significantly increase VCN, without loss of
viability or engraftment (after transplantation). Furthermore, the role
of Rapa on transcriptional and translational events could benefit the
integration of lentiviruses by altering chromosomal accessibility.[78]
Cyclosporine H has recently been shown to efficiently enhance gene
transfer bypassing the innate immune block that can interfere with
efficient gene transfer in HSC. Unlike Cyclosporine A, which inhibits
the host factor CypA, Cyclosporine H inhibits the interferon-induced
transmembrane protein 3 (IFITM3) whose high expression potently
restricts VSV-G pseudotyped LV entry.[79]A
different approach, based on optimization of cell density, was used in
a recent study, which suggested that cell-to-cell contact in a
high-density (4e6/mL) cell culture may mimic bone marrow environment,
allowing more efficient transduction. Ultimately, the combination of
PGE2 and Poloxamer at high-density resulted in an even higher
lentiviral transduction.[80]The
handling of patients' HSC is just as sensitive, if not more, as the
process of making GMP-grade lentiviral particles. Several instruments
and trained personnel are needed to carry out the operations that
involve HSC apheresis, CD34+ selection, transduction, and
cryopreservation in conventional open environments. Even though
individual automated instruments are currently used for each step of
the process, the entire procedure could benefit from a fully automated
closed-system that cuts procedure time, the risk associated with human
error, and costs, which could have more drastic implications once the
drug product reaches commercialization. A
semi-automated immunogenetic selection system, the CliniMACS Plus by
Miltenyi Biotec, is now approved in many countries for autologous CD34+
selection from mobilized apheresis products. The instrument uses a
disposable tubing set that allows all significant steps of processing
to happen in a closed system, thus minimizing the risk of
contamination, a critical factor in cGMP compliance. This method has
proved to be successful at reproducing consistent yields among many
centers.[81] An advanced version of it, the CliniMACS
Prodigy, combines in one instrument the immunoseparation component with
a chamber for further cell manipulation in a closed and fully automated
system. The chamber has a built-in centrifuge and is temperature
controllable from 4C – 37C, making it also an incubator. Briefly,
apheresis products undergo to immunomagnetic selection with
nanobead-conjugated anti CD34 antibodies and buffer purification in a
connected-sterile system with minimum need of operator attendance.[82]
This system is currently used in clinical studies for CAR-T
manufacturing and allows after CD34 selection to perform lentiviral
transduction, further purification and expansion (up to 12 days) within
the same closed system. The end of the process sees the transduced
target cells resuspended into the formulation buffer and potentially
ready to be infused into the patient as drug product.[83]
Summary and Conclusions
At
the moment, the only available curative treatment for patients with BT
and SCD is allogeneic HSCT. HLA matched sibling donor HSCT is now a
well-proven therapy for SCD and BT; however, most patients (>75%) do
not have access to this option due to lack of an available matched
sibling. Alternative donor allogeneic HSCT using unrelated and
haploidentical related donors that would be required for the cure in
most patients with SCD and BTM remain limited by high rates of
transplant-related mortality, GVHD, and graft rejection. Given the
severity of BT and SCD and the shortcomings of current curative
treatment options, BT and SCD remain challenging diseases with a
significant unmet medical need, which is even more aggravated in
countries with poor access to health care. There is a clear need for
potentially curative therapies that can circumvent or eliminate the
deficiencies of current options, and significantly improve associated
morbidity and mortality.
Extensive
progress has been made on gene therapy since its pioneering, making it
a nearly available option for patients. Results from clinical trials of
the last decade indicate that the use of lentiviral vectors can cure
patients affected by hemoglobinopathies. At the moment, in order to
correct the phenotype, many patients require high level of integrations
per genome (~4) in a pancellular fashion. New challenges are
represented by the need to transfer this technology from small to
large-scale volumes, to launch its commercialization. As the medical
field progresses with new and promising pharmacological therapeutic
options, so does the urgency to find better regimens for mobilization
and conditioning of patients for patients that opt to undergo gene
therapy.
Most
importantly, the benefit of the new gene therapy approaches that are
potentially curative would be reinforced if made accessible to a large
number of patients and affordable for the health care system.
References
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167. https://doi.org/10.1016/S0140-6736(17)31822-6
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017;376(16):1561-1573. https://doi.org/10.1056/NEJMra1510865 PMid:28423290
- Piga
A, Perrotta S, Gamberini MR, Voskaridou E, Melpignano A, Filosa A,
Caruso V, Pietrangelo A, Longo F, Tartaglione I, Borgna-Pignatti C,
Zhang X, Laadem A, Sherman ML, Attie KM. Luspatercept improves
hemoglobin levels and blood transfusion requirements in a study of
patients with beta-thalassemia. Blood. 2019;133(12):1279-1289. https://doi.org/10.1182/blood-2018-10-879247 PMid:30617198 PMCid:PMC6440118
- Cappellini
MD, Porter J, Origa R, Forni GL, Voskaridou E, Galacteros F, Taher AT,
Arlet JB, Ribeil JA, Garbowski M, Graziadei G, Brouzes C, Semeraro M,
Laadem A, Miteva D, Zou J, Sung V, Zinger T, Attie KM, Hermine O.
Sotatercept, a novel transforming growth factor beta ligand trap,
improves anemia in beta-thalassemia: a phase II, open-label,
dose-finding study. Haematologica. 2019;104(3):477-484. https://doi.org/10.3324/haematol.2018.198887 PMid:30337358 PMCid:PMC6395345
- Cappellini
MD, Motta I. New therapeutic targets in transfusion-dependent and
-independent thalassemia. Hematology Am Soc Hematol Educ Program.
2017;2017(1):278-283. https://doi.org/10.1182/asheducation-2017.1.278 PMid:29222267 PMCid:PMC6142569
- Lucarelli
G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C,
Politi P, Durazzi SM, Muretto P, Albertini F. Bone marrow
transplantation in patients with thalassemia. N Engl J Med.
1990;322(7):417-421. https://doi.org/10.1056/NEJM199002153220701 PMid:2300104
- Baronciani
D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, Orofino
MG, Giardini C, Al-Ahmari A, Marktel S, de la Fuente J, Ghavamzadeh A,
Hussein AA, Targhetta C, Pilo F, Locatelli F, Dini G, Bader P, Peters
C. Hemopoietic stem cell transplantation in thalassemia: a report from
the European Society for Blood and Bone Marrow Transplantation
Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant.
2016;51(4):536-541. https://doi.org/10.1038/bmt.2015.293 PMid:26752139
- Angelucci
E, Pilo F, Coates TD. Transplantation in thalassemia: Revisiting the
Pesaro risk factors 25 years later. Am J Hematol. 2017;92(5):411-413. https://doi.org/10.1002/ajh.24674 PMid:28181283
- Lucarelli
G, Isgro A, Sodani P, Gaziev J. Hematopoietic stem cell transplantation
in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med.
2012;2(5):a011825. https://doi.org/10.1101/cshperspect.a011825 PMid:22553502 PMCid:PMC3331690
- Li
C, Mathews V, Kim S, George B, Hebert K, Jiang H, Li C, Zhu Y, Keesler
DA, Boelens JJ, Dvorak CC, Agarwal R, Auletta JJ, Goyal RK, Hanna R,
Kasow K, Shenoy S, Smith AR, Walters MC, Eapen M. Related and unrelated
donor transplantation for beta-thalassemia major: results of an
international survey. Blood Adv. 2019;3(17):2562-2570. https://doi.org/10.1182/bloodadvances.2019000291 PMid:31471325 PMCid:PMC6737407
- Zynteglo. European Medicine Agency (EMA). https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo. Accessed October 20, 2019.
- Cavazzana-Calvo
M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady
T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R,
Maouche-Chretien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack
A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie
G, Chretien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros
F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P.
Transfusion independence and HMGA2 activation after gene therapy of
human beta-thalassaemia. Nature. 2010;467(7313):318-322. https://doi.org/10.1038/nature09328 PMid:20844535 PMCid:PMC3355472
- Negre
O, Bartholomae C, Beuzard Y, Cavazzana M, Christiansen L, Courne C,
Deichmann A, Denaro M, de Dreuzy E, Finer M, Fronza R, Gillet-Legrand
B, Joubert C, Kutner R, Leboulch P, Maouche L, Paulard A, Pierciey FJ,
Rothe M, Ryu B, Schmidt M, von Kalle C, Payen E, Veres G. Preclinical
evaluation of efficacy and safety of an improved lentiviral vector for
the treatment of beta-thalassemia and sickle cell disease. Curr Gene
Ther. 2015;15(1):64-81. https://doi.org/10.2174/1566523214666141127095336 PMid:25429463 PMCid:PMC4440358
- Miccio
A, Cesari R, Lotti F, Rossi C, Sanvito F, Ponzoni M, Routledge SJ, Chow
CM, Antoniou MN, Ferrari G. In vivo selection of genetically modified
erythroblastic progenitors leads to long-term correction of
beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(30):10547-10552. https://doi.org/10.1073/pnas.0711666105 PMid:18650378 PMCid:PMC2492493
- Lidonnici
MR, Paleari Y, Tiboni F, Mandelli G, Rossi C, Vezzoli M, Aprile A,
Lederer CW, Ambrosi A, Chanut F, Sanvito F, Calabria A, Poletti V,
Mavilio F, Montini E, Naldini L, Cristofori P, Ferrari G. Multiple
Integrated Non-clinical Studies Predict the Safety of
Lentivirus-Mediated Gene Therapy for beta-Thalassemia. Mol Ther Methods
Clin Dev. 2018;11:9-28. https://doi.org/10.1016/j.omtm.2018.09.001 PMid:30320151 PMCid:PMC6178212
- Boulad
F, Wang X, Qu J, Taylor C, Ferro L, Karponi G, Bartido S, Giardina P,
Heller G, Prockop SE, Maggio A, Sadelain M, Riviere I. Safe
mobilization of CD34+ cells in adults with beta-thalassemia and
validation of effective globin gene transfer for clinical
investigation. Blood. 2014;123(10):1483-1486. https://doi.org/10.1182/blood-2013-06-507178 PMid:24429337 PMCid:PMC3945860
- Romero
Z, Urbinati F, Geiger S, Cooper AR, Wherley J, Kaufman ML, Hollis RP,
de Assin RR, Senadheera S, Sahagian A, Jin X, Gellis A, Wang X,
Gjertson D, Deoliveira S, Kempert P, Shupien S, Abdel-Azim H, Walters
MC, Meiselman HJ, Wenby RB, Gruber T, Marder V, Coates TD, Kohn DB.
beta-globin gene transfer to human bone marrow for sickle cell disease.
J Clin Invest. 2013. https://doi.org/10.1172/JCI67930 PMid:23863630 PMCid:PMC4011030
- Thompson
AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin
E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H,
Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F,
Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarre C, Beuzard Y,
Chretien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C,
Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D,
Petrusich A, Sandler L, Asmal M, Hermine O, De Montalembert M,
Hacein-Bey-Abina S, Blanche S, Leboulch P, Cavazzana M. Gene Therapy in
Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med.
2018;378(16):1479-1493. https://doi.org/10.1056/NEJMoa1705342 PMid:29669226
- Marktel
S, Scaramuzza S, Cicalese MP, Giglio F, Galimberti S, Lidonnici MR,
Calbi V, Assanelli A, Bernardo ME, Rossi C, Calabria A, Milani R,
Gattillo S, Benedicenti F, Spinozzi G, Aprile A, Bergami A, Casiraghi
M, Consiglieri G, Masera N, D'Angelo E, Mirra N, Origa R, Tartaglione
I, Perrotta S, Winter R, Coppola M, Viarengo G, Santoleri L, Graziadei
G, Gabaldo M, Valsecchi MG, Montini E, Naldini L, Cappellini MD, Ciceri
F, Aiuti A, Ferrari G. Intrabone hematopoietic stem cell gene therapy
for adult and pediatric patients affected by transfusion-dependent
ss-thalassemia. Nat Med. 2019;25(2):234-241. https://doi.org/10.1038/s41591-018-0301-6 PMid:30664781
- Bauer
DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC,
Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH,
Stamatoyannopoulos JA, Lettre G, Orkin SH. An erythroid enhancer of
BCL11A subject to genetic variation determines fetal hemoglobin level.
Science. 2013;342(6155):253-257. https://doi.org/10.1126/science.1242088 PMid:24115442 PMCid:PMC4018826
- Bernardo ME, Aiuti A. The Role of Conditioning in Hematopoietic Stem-Cell Gene Therapy. Hum Gene Ther. 2016;27(10):741-748. https://doi.org/10.1089/hum.2016.103 PMid:27530055
- Bacigalupo
A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, Apperley J, Slavin S,
Pasquini M, Sandmaier BM, Barrett J, Blaise D, Lowski R, Horowitz M.
Defining the intensity of conditioning regimens: working definitions.
Biol Blood Marrow Transplant. 2009;15(12):1628-1633. https://doi.org/10.1016/j.bbmt.2009.07.004 PMid:19896087 PMCid:PMC2861656
- Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22(2):53-63. https://doi.org/10.1016/j.blre.2007.10.001 PMid:18039551
- Jacobsohn
DA, Duerst R, Tse W, Kletzel M. Reduced intensity haemopoietic
stem-cell transplantation for treatment of non-malignant diseases in
children. Lancet. 2004;364(9429):156-162. https://doi.org/10.1016/S0140-6736(04)16628-2
- Iannone
R, Casella JF, Fuchs EJ, Chen AR, Jones RJ, Woolfrey A, Amylon M,
Sullivan KM, Storb RF, Walters MC. Results of minimally toxic
nonmyeloablative transplantation in patients with sickle cell anemia
and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519-528. https://doi.org/10.1016/S1083-8791(03)00192-7
- Walters
MC, Hardy K, Edwards S, Adamkiewicz T, Barkovich J, Bernaudin F,
Buchanan GR, Bunin N, Dickerhoff R, Giller R, Haut PR, Horan J, Hsu LL,
Kamani N, Levine JE, Margolis D, Ohene-Frempong K, Patience M,
Redding-Lallinger R, Roberts IA, Rogers ZR, Sanders JE, Scott JP,
Sullivan KM, Multicenter Study of Bone Marrow Transplantation for
Sickle Cell D. Pulmonary, gonadal, and central nervous system status
after bone marrow transplantation for sickle cell disease. Biol Blood
Marrow Transplant. 2010;16(2):263-272. https://doi.org/10.1016/j.bbmt.2009.10.005 PMid:19822218 PMCid:PMC2919571
- Sadelain
M, Riviere I, Wang X, Boulad F, Prockop S, Giardina P, Maggio A,
Galanello R, Locatelli F, Yannaki E. Strategy for a multicenter phase I
clinical trial to evaluate globin gene transfer in beta-thalassemia.
Ann N Y Acad Sci. 2010;1202:52-58. https://doi.org/10.1111/j.1749-6632.2010.05597.x PMid:20712772
- Mansilla-Soto
J, Riviere I, Boulad F, Sadelain M. Cell and Gene Therapy for the
Beta-Thalassemias: Advances and Prospects. Hum Gene Ther.
2016;27(4):295-304. https://doi.org/10.1089/hum.2016.037 PMid:27021486 PMCid:PMC4994056
- Ribeil
JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E,
Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L,
Puy H, Meritet JF, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross
RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S,
Leboulch P, Cavazzana M. Gene Therapy in a Patient with Sickle Cell
Disease. N Engl J Med. 2017;376(9):848-855. https://doi.org/10.1056/NEJMoa1609677 PMid:28249145
- Malik
P, Grimley, M., Quinn, C. T., Shova, A., Little, C., L., Lutzko, C.,
Kalfa, T. A., Niss, O., Mehta, P. A., Chandra, S., Van der Loo, J. C.,
Grassman, E., Witting, S., Nordling, D., Shreshta, A., Felker, S., C.,
Reeves, L., Pillis, D., Loberg, A., Bushman, F. D., Knight-Madden, J.,
Davies, S. M., & Asnani, M. Gene Therapy for Sickle Cell Disease
(SCD) Using RVT-1801 Lentivirus Vector and Arulite Reduced Intensity
Conditioning Transplant Shows Promising Correction of the Disease
Phenotype. American Society of Gene and Cell Therapy; 201; Washington
DC.
- Palchaudhuri
R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, Czechowicz A,
Kfoury Y, Ruchika F, Rossi DJ, Verdine GL, Mansour MK, Scadden DT.
Non-genotoxic conditioning for hematopoietic stem cell transplantation
using a hematopoietic-cell-specific internalizing immunotoxin. Nat
Biotechnol. 2016;34(7):738-745. https://doi.org/10.1038/nbt.3584 PMid:27272386 PMCid:PMC5179034
- Aiuti A, Naldini L. Safer conditioning for blood stem cell transplants. Nat Biotechnol. 2016;3(7):721-723. https://doi.org/10.1038/nbt.3629 PMid:27404882
- Hartigan
AJ, Pearse BR, McDonough SM, Proctor JL, Adams HL, McShea MA, Hoban MD,
Panwar R, Hyzy SL, Goncalves KA, Palchaudhuri R, Boitano AE, Cooke MP.
A Non-Genotoxic Antibody Drug Conjugate Targeting C-Kit for
Hematopoietic Stem Cell Transplant Conditioning. Biology of Blood and
Marrow Transplantation. 2018;24(3):S47-S48. https://doi.org/10.1016/j.bbmt.2017.12.598
- Radtke
S, Adair JE, Giese MA, Chan YY, Norgaard ZK, Enstrom M, Haworth KG,
Schefter LE, Kiem HP. A distinct hematopoietic stem cell population for
rapid multilineage engraftment in nonhuman primates. Sci Transl Med.
2017;9(414). https://doi.org/10.1126/scitranslmed.aan1145 PMid:29093179 PMCid:PMC6467214
- Korbling M, Freireich EJ. Twenty-five years of peripheral blood stem cell transplantation. Blood. 2011;117(24):6411-6416. https://doi.org/10.1182/blood-2010-12-322214 PMid:21460243
- Falzetti
F, Aversa F, Minelli O, Tabilio A. Spontaneous rupture of spleen during
peripheral blood stem-cell mobilisation in a healthy donor. Lancet.
1999;353(9152):555. https://doi.org/10.1016/S0140-6736(99)00268-8
- Becker
PS, Wagle M, Matous S, Swanson RS, Pihan G, Lowry PA, Stewart FM, Heard
SO. Spontaneous splenic rupture following administration of granulocyte
colony-stimulating factor (G-CSF): occurrence in an allogeneic donor of
peripheral blood stem cells. Biol Blood Marrow Transplant.
1997;3(1):45-49.
- Balaguer
H, Galmes A, Ventayol G, Bargay J, Besalduch J. Splenic rupture after
granulocyte-colony-stimulating factor mobilization in a peripheral
blood progenitor cell donor. Transfusion. 2004;44(8):1260-1261. https://doi.org/10.1111/j.1537-2995.2004.00413.x PMid:15265137
- Lindemann
A, Rumberger B. Vascular complications in patients treated with
granulocyte colony-stimulating factor (G-CSF). Eur J Cancer.
1993;29A(16):2338-2339. https://doi.org/10.1016/0959-8049(93)90236-9
- Pilo
F, Angelucci E. Iron Toxicity and Hemopoietic Cell Transplantation:
Time to Change the Paradigm. Mediterr J Hematol Infect Dis. 2019 May
1;11(1):e2019030. doi: 10.4084/MJHID.2019.030. eCollection 2019. https://doi.org/10.4084/mjhid.2019.030 PMid:31205634 PMCid:PMC6548208
- Fitzhugh
CD, Hsieh MM, Bolan CD, Saenz C, Tisdale JF. Granulocyte
colony-stimulating factor (G-CSF) administration in individuals with
sickle cell disease: time for a moratorium? Cytotherapy.
2009;11(4):464-471. https://doi.org/10.1080/14653240902849788 PMid:19513902 PMCid:PMC2747259
- Adler
BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VV, Prchal JT. Fatal
sickle cell crisis after granulocyte colony-stimulating factor
administration. Blood. 2001;97(10):3313-3314. https://doi.org/10.1182/blood.V97.10.3313 PMid:11368061
- De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2(7):581-587. https://doi.org/10.1038/nrd1134 PMid:12815382
- Yannaki
E, Karponi G, Zervou F, Constantinou V, Bouinta A, Tachynopoulou V,
Kotta K, Jonlin E, Papayannopoulou T, Anagnostopoulos A,
Stamatoyannopoulos G. Hematopoietic stem cell mobilization for gene
therapy: superior mobilization by the combination of granulocyte-colony
stimulating factor plus plerixafor in patients with beta-thalassemia
major. Hum Gene Ther. 2013;24(10):852-860. https://doi.org/10.1089/hum.2013.163 PMid:24001178 PMCid:PMC3787462
- Lidonnici
MR, Aprile A, Frittoli MC, Mandelli G, Paleari Y, Spinelli A, Gentner
B, Zambelli M, Parisi C, Bellio L, Cassinerio E, Zanaboni L, Cappellini
MD, Ciceri F, Marktel S, Ferrari G. Plerixafor and G-CSF combination
mobilizes hematopoietic stem and progenitors cells with a distinct
transcriptional profile and a reduced in vivo homing capacity compared
to plerixafor alone. Haematologica. 2017;102(4):e120-e124. https://doi.org/10.3324/haematol.2016.154740 PMid:28034992 PMCid:PMC5395121
- Lagresle-Peyrou
C, Lefrere F, Magrin E, Ribeil JA, Romano O, Weber L, Magnani A, Sadek
H, Plantier C, Gabrion A, Ternaux B, Felix T, Couzin C, Stanislas A,
Treluyer JM, Lamhaut L, Joseph L, Delville M, Miccio A, Andre-Schmutz
I, Cavazzana M. Plerixafor enables safe, rapid, efficient mobilization
of hematopoietic stem cells in sickle cell disease patients after
exchange transfusion. Haematologica. 2018;103(5):778-786. https://doi.org/10.3324/haematol.2017.184788 PMid:29472357 PMCid:PMC5927997
- Boulad
F, Shore T, van Besien K, Minniti C, Barbu-Stevanovic M, Fedus SW,
Perna F, Greenberg J, Guarneri D, Nandi V, Mauguen A, Yazdanbakhsh K,
Sadelain M, Shi PA. Safety and efficacy of plerixafor dose escalation
for the mobilization of CD34(+) hematopoietic progenitor cells in
patients with sickle cell disease: interim results. Haematologica.
2018;103(5):770-777. https://doi.org/10.3324/haematol.2017.187047 PMid:29419425 PMCid:PMC5927989
- Tisdale
JF, Kanter, J., 2, Mapara, M.Y., Kwiatkowski, J.L., Krishnamurti, L.,
Schmidt, M., Miller, A.L., Pierciey, F.J., Shi, W.,7, Ribeil, J.,
Asmal, M., Thompson, Walter, M.C. LentiGlobin Gene Therapy in Patients
with Sickle Cell Disease: Updated Interim Results from HGB-206.
American Society of Gene and Cell Therapy; 2019; Washington D.C.
- Richard
RE, Siritanaratkul N, Jonlin E, Skarpidi E, Heimfeld S, Blau CA.
Collection of blood stem cells from patients with sickle cell anemia.
Blood Cells Mol Dis. 2005;35(3):384-388. https://doi.org/10.1016/j.bcmd.2005.06.014 PMid:16125985
- Uchida
N, Fujita A, Hsieh MM, Bonifacino AC, Krouse AE, Metzger ME, Donahue
RE, Tisdale JF. Bone Marrow as a Hematopoietic Stem Cell Source for
Gene Therapy in Sickle Cell Disease: Evidence from Rhesus and SCD
Patients. Hum Gene Ther Clin Dev. 2017;28(3):136-144. https://doi.org/10.1089/humc.2017.029 PMid:28447889 PMCid:PMC5695729
- Pantin
J, Purev E, Tian X, Cook L, Donohue-Jerussi T, Cho E, Reger R, Hsieh M,
Khuu H, Calandra G, Geller NL, Childs RW. Effect of high-dose
plerixafor on CD34(+) cell mobilization in healthy stem cell donors:
results of a randomized crossover trial. Haematologica.
2017;102(3):600-609. https://doi.org/10.3324/haematol.2016.147132 PMid:27846612 PMCid:PMC5394957
- Ghiaccio
V, Chappell M, Rivella S, Breda L. Gene Therapy for
Beta-Hemoglobinopathies: Milestones, New Therapies and Challenges. Mol
Diagn Ther. 2019;23(2):173-186. https://doi.org/10.1007/s40291-019-00383-4 PMid:30701409
- Hanna
E, Remuzat C, Auquier P, Toumi M. Gene therapies development: slow
progress and promising prospect. J Mark Access Health Policy.
2017;5(1):1265293. https://doi.org/10.1080/20016689.2017.1265293 PMid:28265348 PMCid:PMC5328344
- Barde I, Salmon P, Trono D. Production and titration of lentiviral vectors. Curr Protoc Neurosci. 2010;Chapter 4:Unit 4 21. https://doi.org/10.1002/0471142301.ns0421s53
- Segura
MM, Garnier A, Durocher Y, Ansorge S, Kamen A. New protocol for
lentiviral vector mass production. Methods Mol Biol. 2010;614:39-52. https://doi.org/10.1007/978-1-60761-533-0_2 PMid:20225034
- Valkama
AJ, Leinonen HM, Lipponen EM, Turkki V, Malinen J, Heikura T,
Yla-Herttuala S, Lesch HP. Optimization of lentiviral vector production
for scale-up in fixed-bed bioreactor. Gene Ther. 2018;25(1):39-46. https://doi.org/10.1038/gt.2017.91 PMid:29345252 PMCid:PMC5817386
- Holic
N, Seye AK, Majdoul S, Martin S, Merten OW, Galy A, Fenard D. Influence
of mildly acidic pH conditions on the production of lentiviral and
retroviral vectors. Hum Gene Ther Clin Dev. 2014;25(3):178-185. https://doi.org/10.1089/humc.2014.027 PMid:25073060
- Merten OW, Hebben M, Bovolenta C. Production of lentiviral vectors. Mol Ther Methods Clin Dev. 2016;3:16017. https://doi.org/10.1038/mtm.2016.17 PMid:27110581 PMCid:PMC4830361
- Ansorge
S, Lanthier S, Transfiguracion J, Durocher Y, Henry O, Kamen A.
Development of a scalable process for high-yield lentiviral vector
production by transient transfection of HEK293 suspension cultures. J
Gene Med. 2009;11(10):868-876. https://doi.org/10.1002/jgm.1370 PMid:19618482
- Durocher
Y, Perret S, Kamen A. High-level and high-throughput recombinant
protein production by transient transfection of suspension-growing
human 293-EBNA1 cells. Nucleic Acids Res. 2002;30(2):E9. https://doi.org/10.1093/nar/30.2.e9 PMid:11788735 PMCid:PMC99848
- McCarron
A, Donnelley M, McIntyre C, Parsons D. Transient Lentiviral Vector
Production Using a Packed-Bed Bioreactor System. Hum Gene Ther Methods.
2019;30(3):93-101. https://doi.org/10.1089/hgtb.2019.038 PMid:31084376
- Tinch
S, Szczur K, Swaney W, Reeves L, Witting SR. A Scalable Lentiviral
Vector Production and Purification Method Using Mustang Q
Chromatography and Tangential Flow Filtration. Methods Mol Biol.
2019;1937:135-153. https://doi.org/10.1007/978-1-4939-9065-8_8 PMid:30706394
- Manceur
AP, Kim H, Misic V, Andreev N, Dorion-Thibaudeau J, Lanthier S, Bernier
A, Tremblay S, Gelinas AM, Broussau S, Gilbert R, Ansorge S. Scalable
Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines.
Hum Gene Ther Methods. 2017;28(6):330-339. https://doi.org/10.1089/hgtb.2017.086 PMid:28826344 PMCid:PMC5734158
- Liu
Q. Generation of cGMP-Compliant Stable Packaging and Producer Cell
Lines for Inducible Lentiviral Vector Production. American Society of
Gene and Cell Therapy; 2019; Washington D.C.
- Margherita
Neri FB, Francesca Rossetti, Manuela Cota, Luca Crippa, Silvia Ungari,
Emanuele Simonetti, Luca Allievi, Samuele Corbetta, Federico
Lorenzetti, Francesca Bonfanti, Giuliana Vallanti.
Lentiviral/Retroviral Vector Large Scale Manufacturing. American
Society of Gene and Cell Therapy; 2019; Washington D.C.
- Sutton
RE, Reitsma MJ, Uchida N, Brown PO. Transduction of human progenitor
hematopoietic stem cells by human immunodeficiency virus type 1-based
vectors is cell cycle dependent. J Virol. 1999;73(5):3649-3660.
- Davis
HE, Morgan JR, Yarmush ML. Polybrene increases retrovirus gene transfer
efficiency by enhancing receptor-independent virus adsorption on target
cell membranes. Biophys Chem. 2002;97(2-3):159-172. https://doi.org/10.1016/S0301-4622(02)00057-1
- Davis
HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate
retrovirus transduction via membrane charge neutralization and virus
aggregation. Biophys J. 2004;86(2):1234-1242. https://doi.org/10.1016/S0006-3495(04)74197-1
- Fenard
D, Ingrao D, Seye A, Buisset J, Genries S, Martin S, Kichler A, Galy A.
Vectofusin-1, a new viral entry enhancer, strongly promotes lentiviral
transduction of human hematopoietic stem cells. Mol Ther Nucleic Acids.
2013;2:e90. https://doi.org/10.1038/mtna.2013.17 PMid:23653154 PMCid:PMC4817938
- Lanuti
M, Kouri CE, Force S, Chang M, Amin K, Xu K, Blair I, Kaiser L, Albelda
S. Use of protamine to augment adenovirus-mediated cancer gene therapy.
Gene Ther. 1999;6(9):1600-1610. https://doi.org/10.1038/sj.gt.3300987 PMid:10490770
- Lee
HJ, Lee YS, Kim HS, Kim YK, Kim JH, Jeon SH, Lee HW, Kim S, Miyoshi H,
Chung HM, Kim DK. Retronectin enhances lentivirus-mediated gene
delivery into hematopoietic progenitor cells. Biologicals.
2009;37(4):203-209. https://doi.org/10.1016/j.biologicals.2009.01.008 PMid:19264508
- Delville
M, Soheili T, Bellier F, Durand A, Denis A, Lagresle-Peyrou C,
Cavazzana M, Andre-Schmutz I, Six E. A Nontoxic Transduction Enhancer
Enables Highly Efficient Lentiviral Transduction of Primary Murine T
Cells and Hematopoietic Stem Cells. Mol Ther Methods Clin Dev.
2018;10:341-347. https://doi.org/10.1016/j.omtm.2018.08.002 PMid:30191160 PMCid:PMC6125771
- Hauber
I, Beschorner N, Schrodel S, Chemnitz J, Kroger N, Hauber J, Thirion C.
Improving Lentiviral Transduction of CD34(+) Hematopoietic Stem and
Progenitor Cells. Hum Gene Ther Methods. 2018;29(2):104-113. https://doi.org/10.1089/hgtb.2017.085 PMid:29631437
- Schott
JW, Leon-Rico D, Ferreira CB, Buckland KF, Santilli G, Armant MA,
Schambach A, Cavazza A, Thrasher AJ. Enhancing Lentiviral and
Alpharetroviral Transduction of Human Hematopoietic Stem Cells for
Clinical Application. Mol Ther Methods Clin Dev. 2019;14:134-147. https://doi.org/10.1016/j.omtm.2019.05.015 PMid:31338385 PMCid:PMC6629974
- Heffner
GC, Bonner M, Christiansen L, Pierciey FJ, Campbell D, Smurnyy Y, Zhang
W, Hamel A, Shaw S, Lewis G, Goss KA, Garijo O, Torbett BE, Horton H,
Finer MH, Gregory PD, Veres G. Prostaglandin E2 Increases Lentiviral
Vector Transduction Efficiency of Adult Human Hematopoietic Stem and
Progenitor Cells. Mol Ther. 2018;26(1):320-328. https://doi.org/10.1016/j.ymthe.2017.09.025 PMid:29102562 PMCid:PMC5763075
- Zonari
E, Desantis G, Petrillo C, Boccalatte FE, Lidonnici MR,
Kajaste-Rudnitski A, Aiuti A, Ferrari G, Naldini L, Gentner B.
Efficient Ex Vivo Engineering and Expansion of Highly Purified Human
Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy.
Stem Cell Reports. 2017;8(4):977-990. https://doi.org/10.1016/j.stemcr.2017.02.010 PMid:28330619 PMCid:PMC5390102
- Petrillo
C, Cesana D, Piras F, Bartolaccini S, Naldini L, Montini E,
Kajaste-Rudnitski A. Cyclosporin a and rapamycin relieve distinct
lentiviral restriction blocks in hematopoietic stem and progenitor
cells. Mol Ther. 2015;23(2):352-362. https://doi.org/10.1038/mt.2014.193 PMid:25270076 PMCid:PMC4312494
- Wang
CX, Sather BD, Wang X, Adair J, Khan I, Singh S, Lang S, Adams A,
Curinga G, Kiem HP, Miao CH, Rawlings DJ, Torbett BE. Rapamycin
relieves lentiviral vector transduction resistance in human and mouse
hematopoietic stem cells. Blood. 2014;124(6):913-923. https://doi.org/10.1182/blood-2013-12-546218 PMid:24914132 PMCid:PMC4126331
- Petrillo
C, Thorne LG, Unali G, Schiroli G, Giordano AMS, Piras F, Cuccovillo I,
Petit SJ, Ahsan F, Noursadeghi M, Clare S, Genovese P, Gentner B,
Naldini L, Towers GJ, Kajaste-Rudnitski A. Cyclosporine H Overcomes
Innate Immune Restrictions to Improve Lentiviral Transduction and Gene
Editing In Human Hematopoietic Stem Cells. Cell Stem Cell.
2018;23(6):820-832 e829. https://doi.org/10.1016/j.stem.2018.10.008 PMid:30416070 PMCid:PMC6292841
- Uchida
N, Nassehi T, Drysdale CM, Gamer J, Yapundich M, Demirci S, Haro-Mora
JJ, Leonard A, Hsieh MM, Tisdale JF. High-Efficiency Lentiviral
Transduction of Human CD34(+) Cells in High-Density Culture with
Poloxamer and Prostaglandin E2. Mol Ther Methods Clin Dev.
2019;13:187-196. https://doi.org/10.1016/j.omtm.2019.01.005 PMid:30788387 PMCid:PMC6370599
- Keever-Taylor
CA, Devine SM, Soiffer RJ, Mendizabal A, Carter S, Pasquini MC, Hari
PN, Stein A, Lazarus HM, Linker C, Goldstein SC, Stadtmauer EA,
O'Reilly RJ. Characteristics of CliniMACS(R) System CD34-enriched T
cell-depleted grafts in a multicenter trial for acute myeloid
leukemia-Blood and Marrow Transplant Clinical Trials Network (BMT CTN)
protocol 0303. Biol Blood Marrow Transplant. 2012;18(5):690-697. https://doi.org/10.1016/j.bbmt.2011.08.017 PMid:21875505 PMCid:PMC3762249
- Spohn
G, Wiercinska E, Karpova D, Bunos M, Hummer C, Wingenfeld E, Sorg N,
Poppe C, Huppert V, Stuth J, Reck K, Essl M, Seifried E, Bonig H.
Automated CD34+ cell isolation of peripheral blood stem cell apheresis
product. Cytotherapy. 2015;17(10):1465-1471. https://doi.org/10.1016/j.jcyt.2015.04.005 PMid:25981397
- Aleksandrova
K, Leise J, Priesner C, Melk A, Kubaink F, Abken H, Hombach A, Aktas M,
Essl M, Burger I, Kaiser A, Rauser G, Jurk M, Goudeva L, Glienke W,
Arseniev L, Esser R, Kohl U. Functionality and Cell Senescence of CD4/
CD8-Selected CD20 CAR T Cells Manufactured Using the Automated
CliniMACS Prodigy(R) Platform. Transfus Med Hemother. 2019;46(1):47-54.
https://doi.org/10.1159/000495772 PMid:31244581 PMCid:PMC65