Paul Tieu1, Anthony Chan2,3 and Davide Matino3,4.
1 Department of Pediatrics, McMaster University, Hamilton, Ontario, Canada.
2 Department of Pediatrics, McMaster Children’s Hospital, McMaster University, Hamilton, Ontario, Canada.
3 Thrombosis and Atherosclerosis Research Institute, Hamilton, Ontario, Canada.
4 Department of Medicine, McMaster Children’s Hospital, McMaster University, Hamilton, Ontario, Canada.
Correspondence to: Davide Matino, Department of Medicine, Division of
Hematology & Thromboembolism, McMaster Children’s Hospital, 1280
Main Street West, Hamilton, Ontario. E-mail:
matinod@mcmaster.ca
Published: January 1, 2020
Received: June 6, 2019
Accepted: November 10, 2019
Mediterr J Hematol Infect Dis 2020, 12(1): e2020001 DOI
10.4084/MJHID.2020.001
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
development of neutralizing antibodies in hemophilia is a serious
complication of factor replacement therapy. These antibodies, also
known as “inhibitors”, significantly increase morbidity within the
hemophilia population and lower the quality of life for these patients.
People with severe hemophilia A have an overall 25-40% lifetime risk of
inhibitor development, compared to that of 5-15% lifetime risk in those
with moderate/mild hemophilia A. The risk is lower in hemophilia B
population (about 1-5%) and occurrence of inhibitors is almost only
seen in patients with severe hemophilia B. The understanding of the pathophysiological
mechanism leading to the development of inhibitors in patients with
hemophilia has improved considerably over the last 2 decades.
Identification of early biomarkers which predict inhibitor development
in previously untreated patients with hemophilia will assist in risk
identification and possible early intervention strategies. In this
review, we aim to summarize the molecular mechanisms of inhibitor
development in hemophilia and to identify potential areas in need of
further investigation.
|
Introduction
Hemophilia
A (factor VIII deficiency) is an X-linked, recessive bleeding disorder
due to the deficiency of coagulation factor, and it is estimated to
affect 1 in 5,000 live male births.[1]
Hemophilia A is about four times more common than hemophilia B
(characterized by factor IX deficiency). The severity of the disease is
classified based on the residual amount of functional clotting factor
measured in plasma, with persons with <1% factor defined as severe;
1-5% as moderate; and >5%-<40%, as mild.[2]
Although clinical trials involving gene therapy are currently ongoing,
there is no available cure for hemophilia yet. Current treatments
require lifelong, frequent, intravenous infusions of expensive clotting
factor protein that are manufactured from human plasma or through
recombinant DNA technology.
Moreover, about 30% of severe
hemophilia A patients and 5% of severe hemophilia B patients on
replacement therapy develop an immune response to the exogenous
protein. The development of neutralizing antibodies in hemophilia is a
severe complication of factor replacement therapy. Antibodies that
neutralize the procoagulant function of factors are known as
inhibitors. The incidence of inhibitor development reflects the
severity of the molecular defect: FVIII inhibitors develop in 20% to
35% of patients with severe hemophilia A and in 3% to 13% of
mild/moderate patients.[3-5] Immune tolerance to
factors has been a major concern and interest for many years because
the development of inhibitors significantly increase morbidity and
lower the quality of life within the hemophilia population. While
hematologists and immunologists have developed and tested a myriad of
different drugs and techniques in animal model of hemophilia, current
treatments available to by-pass inhibitors in patients are few,
variable in their effectiveness, and extremely expensive.[6]
Different risk factors have been proposed to be associated with
inhibitor development. These include risk factors associated with the
type of preparation of therapeutic FVIII (i.e., either the plasmatic or
recombinant origin of FVIII), with the inflammatory state or the HLA
haplotype of the patient, or with polymorphisms in immune genes such as
genes encoding tumor-necrosis factor, interleukin-10, or CTLA-4.[7-9]
However, the only proven risk factor is the type of mutation in the F8
gene that causes hemophilia A, and more specifically the presence or
absence of traces of endogenous FVIII antigen in the circulation of the
patient. Indeed, in a mouse model of hemophilia A, FVIII mRNA has been
detected in mouse thymus, and intrathymic injection of FVIII into
neonatal FVIII knockout mice generates tolerance to subsequent
immunization with FVIII.[10,11] These findings strongly suggest that T and B cells reactive to FVIII are deleted through central tolerance mechanisms.
The
understanding of the pathophysiological mechanism leading to the
development of inhibitors in patients with hemophilia has improved
considerably over the last two decades. This process is complex and
involves cells, cytokines, and other immune regulatory molecules. This
review aims to summarize our current understanding of the molecular
mechanisms that lead to inhibitor synthesis and potential areas in need
of further investigation.
Primary Immune Response
Factor endocytosis by APCs and presentation to T-cell.
Understanding the location where therapeutic factors encounter the
immune system for the first time, the type of antigen presenting cells
that are involved in the process and the site where the anti-factor
immune response develops is crucial for developing strategies to
selectively prevent the onset of the deleterious anti-FVIII and
anti-FIX immune response. The first encounter of the infused factor
with immune effectors most likely occurs in the spleen.
Blood-borne antigens reach the spleen through the splenic artery, which
branches either towards the red pulp and interacts with red pulp
macrophages or towards the marginal zone of the spleen, which contains
three major types of professional APCs: macrophages, B lymphocytes and
dendritic cells.[12,13] This view is supported by the work of Navarette et al.[14]
where they demonstrated that human FVIII administered to
FVIII-deficient mice preferentially accumulates in the marginal zone
(MZ) of the spleen. The disruption of splenic germinal centers by
intravenous injection of anti-CD154 antibodies also caused a reduction
in anti-FVIII antibody titers and abolition of T-cell responses to
FVIII.[15] Therefore, identification of the receptors
implicated in retention of therapeutic factors in the marginal zone may
contribute towards novel strategies aimed at reducing their
immunogenicity. In addition, the removal of the spleen or selective in
vivo depletion of APCs before repeated FVIII administration reduces the
extent of the anti-FVIII immune response.[14]
Interestingly, the development of detectable anti-FVIII immune response
to therapeutic FVIII was observed in splenectomized animals, indicating
that alternative secondary organs, the lymph nodes or possibly the bone
marrow, may be involved in the immune response to therapeutic factors
as well.[16] On the other hand, another hypothesis is
that since bleeding and coagulation create a highly inflammatory
microenvironment, therapeutic FVIII/FIX may be captured by
antigen-presenting cells at the site of bleeding and then transported
to secondary lymphoid organs for presentation to naïve CD4+ T cells.
The inflammatory atmosphere could attract locally cells of innate
immunity and antigen-presenting cells. The environment may also provide
the appropriate signals for the activation of the professional
antigen-presenting cells that have endocytosed FVIII and processed
FVIII into peptides, about 9-14 amino acids in lengths.[17]
FVIII-educated APCs likely migrate to the secondary lymphoid organs
which are rich in T-cell like the periarteriolar lymphoid sheath
surrounding the splenic artery. There, mature APCs are surveyed by CD4+
T cells that express T cell receptors specific for FVIII peptides bound
to MHC class II molecules.
Different types of APCs may be involved
in the uptake of therapeutic FVIII in patients. Among these, dendritic
cells, macrophages, and B lymphocytes are the most potent. However, the
types of APCs differ depending on the “experience” the immune system of
the patient has, of exogenous FVIII. In untreated patients who have
never been exposed to FVIII, FVIII-specific B lymphocytes have not been
triggered and are not likely to be present at a frequency high enough
to serve as APCs. B cells and macrophages, although considered
professional antigen-presenting cells, most likely do not present FVIII
to naıve CD4+ T cells because of the high specificity and strength of
immune synapse formation required to activate naıve CD4+ T cells.[18]
Therefore, in view of the capacity to stimulate naïve T cells, DCs are
likely to be the major APC involved in the primary immune response to
clotting factors. DCs are derived from bone marrow and circulate as
precursors in blood before entering tissues where they become resident
immature DCs that can sense changes in their local environment.[19]
Immature DCs can take up antigen using both receptor- and
non-receptor-mediated mechanisms and degrade antigens in endocytic
vesicles to produce antigenic peptides capable of binding to MHC-class
II.[19] Maturation of DCs requires danger signals
provided by exogenous or endogenous stimuli such as pathogen-derived
products, inflammatory cytokines, or CD40-CD40 ligand interactions. As
DCs mature, they express a high density of MHC-class II molecules
complexed with antigenic peptides and upregulate costimulatory
molecules. Antigenic peptides complexed with MHC-class II are
recognized by the T-cell receptor (TCR) expressed on CD4+ T cells. When
human dendritic cells are cultured with FVIII in vitro, this does not
lead to DC maturation.[20] The authors concluded that
FVIII does not possess inherent danger signals for human DCs. However,
certain FVIII products that might have undergone inappropriate
production procedures could develop inherent danger signals for the
immune system.[21,22] In addition, the monocyte
derived DCs used in this study may not be representative of the entire
DC population in the body. The causative factors for this difference in
the in vitro and in vivo recognition of FVIII by the immune system
remains unclear, but, likely, the microenvironment within which FVIII
is taken up and presented by immune cells plays an important role in
this response.[20,23]
Several
endocytic receptors specific for FVIII have been characterized. Members
of the low-density lipoprotein receptor (LDLR) family recognize protein
structures in the heavy and light chains of FVIII 70}.[24,25] Asialoglycoprotein receptor binds to galactose-ending glycans of the B domain of FVIII.[26] The macrophage mannose receptor (MMR/CD206) interacts with mannose-ending glycans on the A1 and C1 domains of the molecule.[27] Dasgupta et al.[27]
used human monocyte-derived dendritic cells to demonstrate that FVIII
is endocytosed by the macrophage mannose receptor (CD206) that
recognizes mannose-ending glycans on both the heavy and light chains of
FVIII. Mechanistically, VWF has been shown to prevent the binding of
FVIII to macrophage mannose receptor and block the endocytosis of FVIII
by monocyte derived dendritic cells in a dose-dependent manner.[27,28] Therefore, VWF has been proposed to reduce the immunogenicity of FVIII in patients with hemophilia A.[29,30]
However, in recent studies, the blockage of the mannose receptors by
mannan did not produce the expected effect in reducing uptake by
dendritic cells, suggesting that additional, as yet unidentified,
endocytic receptors are of clinical significance.[31,32]
On the other hand, the monoclonal antibody KM33 targets the FVIII C1
domain, specifically residues Arg2090, Lys2092, and Phe2093.[33,34]
It has been shown to completely inhibit FVIII endocytosis by both
monocyte-derived dendritic cells and bone marrow-derived dendritic
cells by targeting an epitope of FVIII that is essential for its
uptake. Specifically, KM33 interferes with the binding of FVIII to
low-density lipoprotein receptor–related protein-1 (LRP) and dendritic
cell-specific intercellular adhesion molecule-3-grabbing non-integrin
(DC-SIGN) receptors.[32] In vivo administration of KM33 significantly reduced the production of neutralizing antibodies against FVIII.[32]
The in vitro and in vivo inhibitory effect of KM33 suggests that these
interactive surfaces on the FVIII C1 domain are critical for the
initiation of immune response to therapeutic FVIII.
Moreover,
infusions of FVIII variant proteins with alanine substitutions at the
positions Arg2090, Lys2092, and Phe2093 in FVIII-deficient mice led to
reduced T-cell and B-cell responses as compared with wild-type FVIII.[34]
Therapeutic monoclonal antibodies to inflammatory cytokines or
immunosuppressive agents such as steroids have been shown to limit the
activation state and endocytic capacity of APCs.[35,36]
Therefore, the inflammatory environment of the patients could be
neutralized before or at the time of administration of therapeutic
clotting factors. Besides, high-intensity FVIII treatment because of
excessive bleeding episodes may allow FVIII to compete more efficiently
with other antigens for uptake by APCs, resulting in more efficient
presentation of FVIII-derived peptides to CD4+ T cells.[30] As a result, high-intensity FVIII treatment has been linked to higher inhibitor development.[37]
Dendritic
cells endocytose and process therapeutic clotting factors into
peptides, which are loaded onto the cleft of MHC-II molecules and
expressed on the surface of the dendritic cell.[17]
During dendritic cell maturation, they also express co-stimulatory
molecules such as CD80/86 and CD40 needed for CD4+ T cell activation.[23,38]
In the secondary lymphoid organs, mature dendritic cells are surveyed
by FVIII-specific CD4+ T cells until cognate MHCII-TCR interactions are
established; the engagement of co-stimulatory molecules between the
dendritic cell and T cell (i.e., CD40 with CD40L, CD80/CD86 with CD28)
occurred; and cytokine secretion by both the dendritic cell and T cell
happened to induce T cell activation and proliferation.[39]
Several novel strategies have been developed from the understanding of
this interactive mechanism. For instance, the abrogation of the
cross-talk between APCs and T cells using anti-CD40L monoclonal
antibody or CTLA4-Ig constructs showed promising results in
FVIII-deficient mice.[15,40] In
naïve animals, the use of blocking antibodies to disrupt the cognate
interaction between T cells and APCs caused immunological
hyporesponsiveness to FVIII, or the partial breakdown of an immune
response in FVIII-primed mice.[15,40-42] In humans, only three hemophilia A patients with FVIII inhibitors (> 10 BU/ml) have been treated with anti-CD40L.[43]
Inhibitor levels were reported to decrease in these patients. However,
more evidence suggested that treatment with anti-CD40L was associated
with both arterial and venous thromboembolic complications.[44,45]
Mechanistically, CD40 and CD40L are both expressed on platelets, and
the use of an anti-CD40 antibody can activate platelets, thus
increasing the likelihood of thrombotic events. Therefore, CD40-CD40L
blockade cannot be considered as a safe alternative for FVIII tolerance
induction at the moment.[39]
T-cell presentation to B-cell and B-cell proliferation.
Activated CD4+ T cells trafficke to the B cell follicles in the spleen
where they activate FVIII specific naïve B cells. Bone marrow derived B
cells internalize FVIII via receptor-mediated endocytosis with
FVIII-specific membrane-tethered immunoglobulin and interact with
activated CD4+ T cells via an MHC II-TCR association.[46]
Activated B cells then proliferate and terminally differentiate into
FVIII-specific memory B cells or anti-FVIII antibody secreting plasma
cells. Memory B cells do not secrete anti-FVIII antibodies. These cells
reside in the spleen or bone marrow and quickly terminally
differentiate into plasma cells after subsequent exposure to FVIII.[39]
Meanwhile,
plasma cells can be either short-lived or, depending on survival
factors present during their development, they can reside in the spleen
or the bone marrow as long-lived cells.[47,48] In
fact, FVIII-specific plasma cells have been demonstrated to survive for
a very long time in the absence of further FVIII immunizations in mice.[49]
In naïve mice, anti-CD40L blocks the germinal center reaction by
preventing cognate T cell-B cell interactions. This would stop the
production of new plasma cells and lead to a reduction in the levels of
circulating anti-FVIII antibodies in the plasma over time as
short-lived plasma cells senesced. However, long-lived plasma cells,
which no longer require significant T cell costimulation, could occupy
survival niches in the spleen and bone marrow and continue to maintain
some level of anti-FVIII Ab production.[39] Strategies to modulate the primary immune response in hemophilia are summarized in Figure 1.
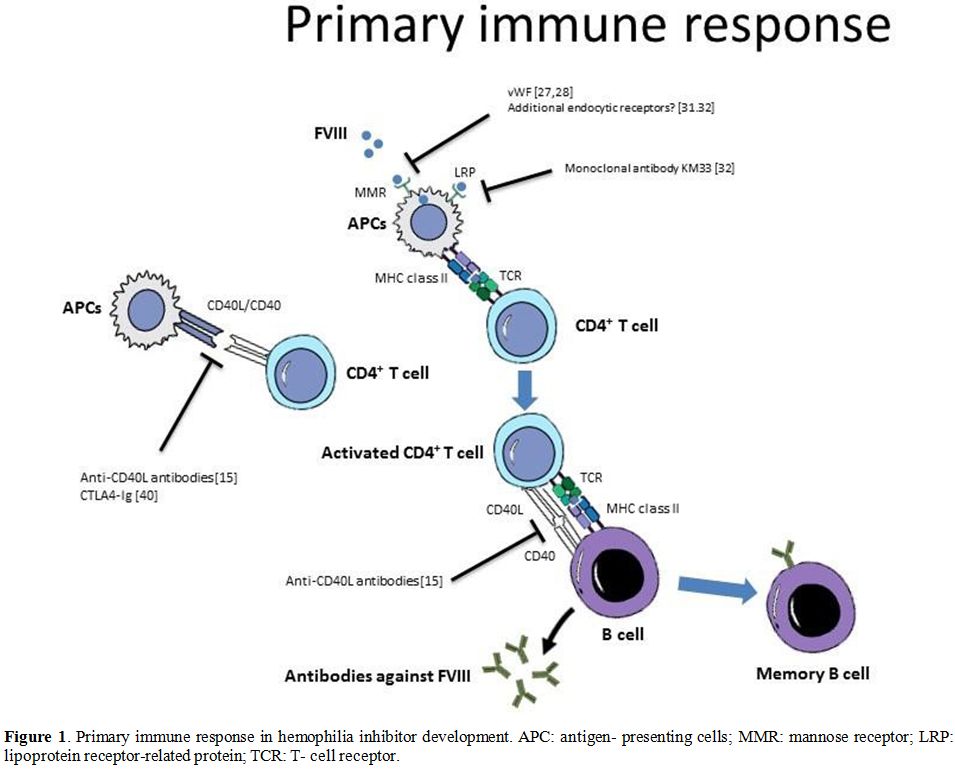 |
Figure
1. Primary immune response in hemophilia inhibitor development. APC:
antigen- presenting cells; MMR: mannose receptor; LRP: lipoprotein
receptor-related protein; TCR: T- cell receptor. |
Secondary Immune Response
During
the secondary immune response, FVIII-specific memory B cells generated
during the primary immune response act as APCs and activate
FVIII-specific CD4+ T cells. With the help of CD4+ T cells,
FVIII-specific memory B cells further differentiate into ASCs.
Meanwhile, uptake of FVIII by other professional APCs, such as the
dendritic cells, results in activation of T cells that, in turn,
activate new FVIII-specific B cells and thus generate additional ASCs
and memory B cells. Several studies investigating the mechanisms of
immune tolerance induction demonstrated that high FVIII levels might
inhibit memory B cell differentiation.[50,51] Indeed, Reipert et al.[52]
discovered that high FVIII concentration could inhibit FVIII-specific
memory B cells both in vitro and in vivo. In these studies, splenocytes
(depleted of CD138+ plasma cells) were obtained from mice that were
repeatedly immunized with FVIII. This CD138- splenocyte pool,
therefore, represented a population of memory B cells, which was
restimulated in vitro or in vivo, using an adoptive transfer model with
increasing concentrations of FVIII. When CD138- splenocytes were
restimulated with supraphysiological concentrations of FVIII (between 1
and 20 mcg/mL), potentially mirroring the FVIII levels in some
high-dose ITI patients, this memory cell population was incapable of
differentiating into anti-FVIII Ab secreting plasma cells. In contrast,
physiological FVIII concentrations (0.01–0.1 mcg/mL) supported memory B
cell differentiation.
Moreover, Matino et al.[53]
demonstrated that induced CD4+FOXP3+ cells were capable of suppressing
the differentiation of FVIII-specific memory B cells into FVIII
antibody–producing plasma cells in vitro. On the other hand, most
antibodies secreted from the plasma cells are mainly of the
immunoglobulin IgG1 and IgG4 subtypes and directed against the A2
and/or C2 domains of FVIII. Several epitopes of both neutralizing and
non-neutralizing types located outside these, some in the B domain,
have also been described.[54,55] The main mechanism
by which the antibodies neutralize the factor is by steric hindrance,
but the formation of immune complexes and subsequently, the enhanced
catabolism as well as hydrolysis have also been suggested.[56] They can interfere with FVIII binding to phospholipids or VWF via binding to the C2 domain.[57,58]
Besides, the antibodies can interfere with FVIII binding to FIX or
block the intrinsic X-ase activity of the VIIIa-IXa complex.[59,60] Alternatively, the antibodies can increase clearance of VIII via direct proteolysis.[56,61]
Regarding non neutralizing antibodies, it remains debated as to whether
these antibodies or at least any immune response they provoke, are of
clinical significance and should be considered as well.[62-64] Strategies to modulate the secondary immune response in hemophilia are summarized in Figure 2.
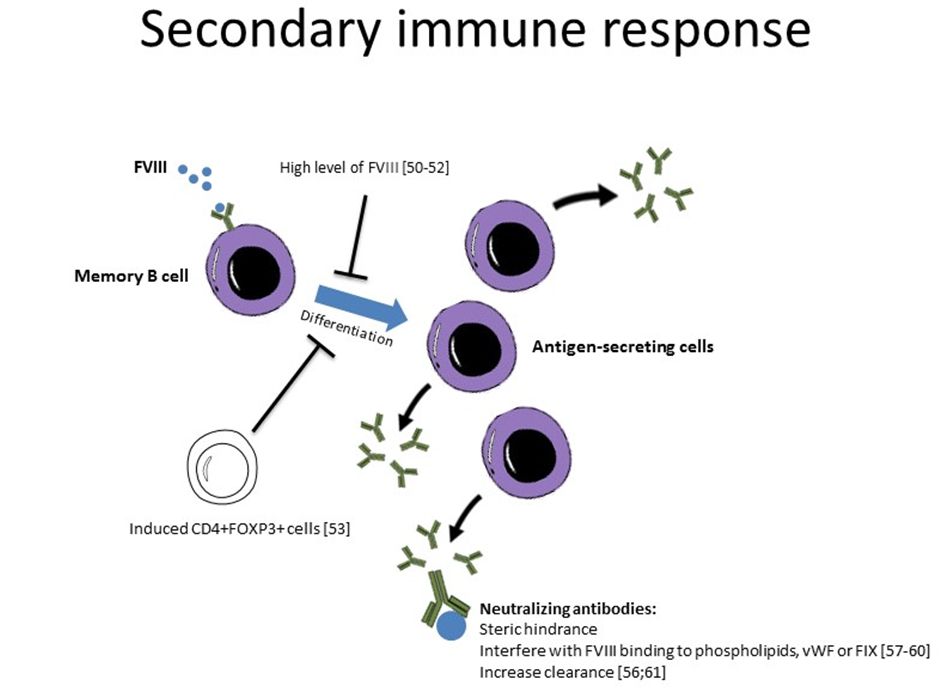 |
Figure 2 |
Actors in Inhibitor Development
Inhibitors are high-affinity antibodies. They are primarily immunoglobulin G (IgG) directed against the factor protein.[65]
Inhibitors in individuals with acquired hemophilia are often
monoclonal. In one study, approximately 80% of individuals with
hemophilia A who developed inhibitors had at least two or more
independent antibody specificities against factor VIII.[66]
There is a distinct spectrum of neutralizing and non-neutralizing
antibodies in different cohorts of patients with severe hemophilia A
and in healthy individuals.[67] IgG4 and IgG1 were
the most abundant IgG subclasses in patients with FVIII inhibitors,
while IgG4 was utterly absent in patients without FVIII inhibitors and
in healthy subjects.[67] In addition, FVIII-specific
antibodies in hemophilia A patients with inhibitors have approximately
100-fold higher apparent affinities than that of antibodies found in
patients without inhibitors or in healthy individuals.[65]
In patients who are never exposed to the deficient factor, the immune
response presumably takes place by dendritic cell pathways, whereas
among primed patients with an established immune response, the B cells
seem to be the key APCs.[68] The importance of
cross-talk between APC and CD4+ T cells has been shown in animal models
using antibodies toward costimulatory cell surface molecules
interfering with the binding to the CD40 ligand, CD80/86, and CTLA4.[40-42,51,69,70]
Indirect evidence of the role that CD4+ cells play in anti-FVIII
antibody synthesis comes from the observation that inhibitors may
spontaneously disappear in conjunction with an HIV-associated decline
in CD4+ counts.[71] More recently, the prevention of
inhibitor synthesis in a murine haemophilia model by blockade of
costimulatory signals has provided direct evidence that CD4+ cells are
indeed essential for the development of an anti-FVIII antibody
response.[40] Besides, for the CD4+ T cells to become
activated and acquire the capacity to stimulate antigen-specific B-cell
differentiation into antibody-secreting plasma cells, additional
triggers or alert signals are often required, as suggested in the
danger model theory.[72] These danger signals are
mainly released by cell death, tissue damage, stress, and systemic
inflammatory responses, e.g., interleukins (ILs), heat shock proteins,
adenosine triphosphate, reactive oxygen species, and growth factors.[73]
Whether a T cell-independent immune response toward FVIII is evoked
into producing FVIII-specific antibodies is not completely clear, but
this could potentially be of relevance for the formation of
non-neutralizing antibodies and/or low-affinity antibodies.[74]
Following antigenic stimulation, naive CD4+ cells may differentiate
into one of several T-cell subsets that differ in function and cytokine
secretion. Th1 cells secrete pro-inflammatory cytokines such as IL-2
and IFN-γ and help in the synthesis of complement-fixing antibodies
such as IgG1.[75]
On the other hand, Th2 cells
can have a down-regulatory effect on the immune response by secreting
anti-inflammatory cytokines such as IL-4 and IL-10, which inhibit the
proliferation and function of Th1 cells and antigen-presenting cells.
However, Th2 cells can also stimulate B cells that produce certain
antibody subclasses such as IgG4. In fact, high-affinity FVIII-specific
antibodies found in patients with FVIII inhibitors are predominantly
IgG4. This suggests a distinct immune regulatory pathway responsible
for the development of FVIII-specific IgG4 associated with FVIII
inhibitors.[52,67] Overall, inhibitor production by B cells is controlled by a complex interaction of different CD4+ subsets.[75] Reding et al.[76]
demonstrated the importance of both Th1 and Th2 cells in the synthesis
of anti-FVIII antibodies. More intense anti-FVIII antibody responses
and higher inhibitor titres correlate with a predominance of Th2-driven
IgG4. Successful immune tolerance therapy in haemophilia A patients and
immunosuppressive therapy in acquired haemophilia patients correlate
with a predominance of Th1-driven anti-FVIII antibody.[1]
To
further define the role of T cells in the pathogenesis of FVIII
inhibitors, Reding and colleagues mapped the CD4+ T-cell epitopes on
FVIII.[77,78] They found three immunodominant CD4+
epitopes on the FVIII C2 domain, corresponding to residues 2191–2210,
2241–2290, and 2291–2330.[77] Each of these epitopes
overlaps inhibitor-binding sites, suggesting that CD4+ cells
recognizing these sequences may be involved in the regulation of
inhibitor synthesis. Besides, there is a lack of recognition of
specific CD4+ epitopes correlated with inhibitor formation.[77]
For instance, the absence of recognition of residues 2191–2210
correlates with inhibitor formation, suggesting that a pathogenic
immune response to FVIII results from failure to activate regulatory
CD4+ cells specific for certain FVIII sequences. On the other hand,
Reding and colleagues found notable differences between the CD4+
epitope repertoires of congenital and acquired haemophilia patients.
This suggests different mechanisms of inhibitor formation, which is
expected, given that inhibitors are a consequence of an alloimmune
response in congenital haemophilia A patients and an autoimmune
response in acquired haemophilia patients.
Tregs have also been
implicated in the process of inducing tolerance in patients with an
established memory using immune tolerance induction therapy. Frequent
exposure to the deficient factor in the absence of systemic
inflammation may induce Tregs with a subsequent lack of T-helper cells,
preventing B-cell differentiation and promoting tolerance through
B-cell anergy and/or deletion.[79] High doses in a
murine model of hemophilia A irreversibly inhibited the memory B cells
via an indirect effect on both APCs and T cells.[50]
The importance of T-regulatory cells in the process of antibody
formation has been established, and to date, different subsets of cells
with suppressor activities have been defined.[80]
Notably, the CD4+CD25+FoxP3+ Treg cells have been well-studied. They
originate during thymic T-cell development and are also referred to as
natural Tregs.[3] They may also be induced in the
periphery from conventional T cells. Treg activation occurs through
antigen-specific binding to T-cell receptors, but the suppression
appears to be a more nonspecific event, which may add somewhat to the
complexity of inhibitor formation. The action of Tregs is
multifactorial and includes direct cell contact-dependent mechanisms
involving APCs and/or effector T cells, as well as cytokine-mediated
suppression of proliferation and differentiation. Tregs may also
promote the secretion of suppressive factors by dendritic cells.[81]
Moreover,
indoleamine 2,3-dioxygenase 1 (IDO1) is a key regulatory enzyme that
supports Treg function and peripheral tolerance in adult life. Matino
et al.[53] discovered in both human and hemophilic
mouse that defective TLR9-mediated activation of IDO1 induction was
associated with an inhibitor-positive status. These findings indicate
the novel strategies of improving the IDO1 function in preventing or
eradicating inhibitors to therapeutic administered FVIII.[53]
Factor IX Inhibitors
Mechanistic
studies on inhibitor development in hemophilia B have been studied
extensively compared with hemophilia B. Hemophilia A is four times as
frequent as hemophilia B, and the incidence of inhibitors is higher.[1]
Further, hemophilia B is often associated with point mutations, which
are less commonly associated with inhibitor development, rather than
deletions. The extent to which the mechanistic information from
hemophilia A can be generalized to hemophilia B is not known and may
differ substantially. While the clinical phenotype of haemophilia B is
indistinguishable from that of haemophilia A, there are clear
differences regarding inhibitor development between the two conditions.
The development of FIX inhibitors is much less common than in
hemophiliia A, occurring in approximately 5% of those with severe
hemophilia B.[82] The majority of those affected
(approximately 80%) are high responders, and 50% or more have a history
of severe allergic reactions to FIX products.[82]
Although the development of pathogenic immune responses against FIX is
less common, induction of immune tolerance to FIX is not often
successful, occuring in only approximately 15% of treated patients in
most series.[82] However, the mechanisms of the
immune response to FIX replacement therapy in humans have not been well
studied and are thus poorly understood. More work in this area is
needed.
Conclusions
The
purpose of this review was to summarize the molecular mechanisms of
inhibitor development in hemophilia and to identify potential areas in
need of further investigation. Understanding the location where
therapeutic factors encounter the immune system for the first time, and
the site where the anti-factor immune response develops is essential
for developing novel strategies towards immune tolerance. Previous work
targeting the primary immune response in the splenic germinal centers
by anti-CD154 antibodies showed promising results in hemophilia A.[15]
Besides the spleen, alternative secondary organs, including the lymph
nodes or possibly the bone marrow, may be involved in the immune
response to therapeutic factors as well.[16] In view
of the capacity to stimulate naïve T cells, dendritic cells are likely
to be the major antigen-presenting cells involved in the primary immune
response to clotting factors. However, FVIII might not possess inherent
danger signals for human dendritic cells. Pfistershammer et al.[20]
demonstrated that when human dendritic cells are cultured with FVIII in
vitro, this does not lead to DC maturation. The causative factors for
this difference in the in vitro and in vivo recognition of FVIII by the
immune system remains unclear, but, likely, the microenvironment within
which FVIII is taken up and presented by immune cells plays an
essential role in this response.[20,23]
On the other hand, several endocytic receptors specific for FVIII have
been characterized and they can be the potential targets to reduce the
immunogenicity of therapeutic factors. For example, VWF has been shown
to prevent the binding of FVIII to macrophage mannose receptor and
block the endocytosis of FVIII by monocyte derived dendritic cells in a
dose-dependent manner.[27,28] In addition, the
monoclonal antibody KM33, which targets an epitope of FVIII, has been
shown to completely inhibit FVIII endocytosis by dendritic cells. In
the secondary lymphoid organs, the engagement of co-stimulatory
molecules between the mature dendritic cell and T cell (i.e. CD40 with
CD40L, CD80/CD86 with CD28) occurred. A novel treatment using
anti-CD40L had been employed in three hemophilia A patients with
inhibitors.[43] Although inhibitor levels decreased
in these patients, treatment with anti-CD40L was associated with both
arterial and venous thromboembolic complications.[44,45]
Activated CD4+ T cells trafficke to the B cell follicles in the spleen,
where they activate FVIII specific naïve B cells. Activated B cells
then proliferate and terminally differentiate into FVIII-specific
memory B cells or anti-FVIII antibody secreting plasma cells. Naïve
mice treated with anti-CD40L appeared to have the production of new
plasma cells stopped, which eventually led to a reduction in the levels
of circulating anti-FVIII antibodies in the plasma over time as
short-lived plasma cells senesced. During the secondary immune
response, FVIII-specific memory B cells further differentiate into
antibody-secreting cells. Antibodies neutralize the therapeutic factor
in different ways. They can interfere with FVIII binding to
phospholipids or VWF via binding to the C2 domain.[57,58] They can interfere with FVIII binding to FIX or block the intrinsic X-ase activity of the VIIIa-IXa complex.[59,60] Alternatively, the antibodies can increase clearance of VIII via direct proteolysis.[56,61]
Several studies investigating the mechanisms of immune tolerance
induction demonstrated that high FVIII levels might inhibit memory B
cell differentiation.[50,51]Regarding
nonneutralizing antibodies, it remains debated as to whether these
antibodies, or at least any immune response they provoke, are of
clinical significance and should be considered as well.[62-64]
In addition, high-affinity FVIII-specific antibodies found in patients
with FVIII inhibitors are predominantly IgG4, and that suggests a
distinct immune regulatory pathway responsible for the development of
FVIII-specific IgG4 associated with FVIII inhibitors.[52,67]
Overall, the prevention of antibody development against FVIII during
replacement therapy of patients with hemophilia A remains a major goal
in the design of future treatment strategies. Identification of early
biomarkers that predict inhibitor development in previously untreated
patients with hemophilia A will assist in risk identification and
possible early intervention strategies. In the last decade, advances
have been made in our understanding of the mechanism of the immune
response to therapeutic factors in hemophilia patients. A clear
understanding of the relevance of these mechanisms in the context of
successful immune tolerance therapy, and ultimately gene therapy,
awaits further study.
References
- Soucie JM, Evatt B, Jackson D. Occurrence of
hemophilia in the United States. The Hemophilia Surveillance System
Project Investigators. Am J Hematol 1998; 59: 288-294. https://doi.org/10.1002/(SICI)1096-8652(199812)59:4<288::AID-AJH4>3.0.CO;2-I
- Blanchette
VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A.
Definitions in hemophilia: communication from the SSC of the ISTH. J
Thromb Haemost 2014; 12: 1935-1939. https://doi.org/10.1111/jth.12672 PMid:25059285
- Antonarakis
SE, Rossiter JP, Young M, Horst J, de MP, Sommer SS, Ketterling RP,
Kazazian HH, Jr., Negrier C, Vinciguerra C, Gitschier J, Goossens M,
Girodon E, Ghanem N, Plassa F, Lavergne JM, Vidaud M, Costa JM, Laurian
Y, Lin SW, Lin SR, Shen MC, Lillicrap D, Taylor SA, Windsor S, Valleix
SV, Nafa K, Sultan Y, Delpech M, Vnencak-Jones CL, Phillips JA, III,
Ljung RC, Koumbarelis E, Gialeraki A, Mandalaki T, Jenkins PV, Collins
PW, Pasi KJ, Goodeve A, Peake I, Preston FE, Schwartz M, Scheibel E,
Ingerslev J, Cooper DN, Millar DS, Kakkar VV, Giannelli F, Naylor JA,
Tizzano EF, Baiget M, Domenech M, Altisent C, Tusell J, Beneyto M,
Lorenzo JI, Gaucher C, Mazurier C, Peerlinck K, Matthijs G, Cassiman
JJ, Vermylen J, Mori PG, Acquila M, Caprino D, Inaba H. Factor VIII
gene inversions in severe hemophilia A: results of an international
consortium study. Blood 1995; 86: 2206-2212. https://doi.org/10.1182/blood.V86.6.2206.bloodjournal8662206 PMid:7662970
- Schwaab
R, Brackmann HH, Meyer C, Seehafer J, Kirchgesser M, Haack A, Olek K,
Tuddenham EG, Oldenburg J. Haemophilia A: mutation type determines risk
of inhibitor formation. Thromb Haemost 1995; 74: 1402-1406. https://doi.org/10.1055/s-0038-1649954 PMid:8772209
- Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia 1998; 4: 558-563. https://doi.org/10.1046/j.1365-2516.1998.440558.x PMid:9873794
- Gomez
K, Klamroth R, Mahlangu J, Mancuso ME, Mingot ME, Ozelo MC. Key issues
in inhibitor management in patients with haemophilia. Blood Transfus
2014; 12 Suppl 1: s319-s329.
- Astermark
J, Oldenburg J, Carlson J, Pavlova A, Kavakli K, Berntorp E, Lefvert
AK. Polymorphisms in the TNFA gene and the risk of inhibitor
development in patients with hemophilia A. Blood 2006; 108: 3739-3745. https://doi.org/10.1182/blood-2006-05-024711 PMid:16926287
- Astermark
J, Oldenburg J, Pavlova A, Berntorp E, Lefvert AK. Polymorphisms in the
IL10 but not in the IL1beta and IL4 genes are associated with inhibitor
development in patients with hemophilia A. Blood 2006; 107: 3167-3172. https://doi.org/10.1182/blood-2005-09-3918 PMid:16380445
- Astermark
J, Wang X, Oldenburg J, Berntorp E, Lefvert AK. Polymorphisms in the
CTLA-4 gene and inhibitor development in patients with severe
hemophilia A. J Thromb Haemost 2007; 5: 263-265. https://doi.org/10.1111/j.1538-7836.2007.02290.x PMid:17269936
- Hollestelle
MJ, Thinnes T, Crain K, Stiko A, Kruijt JK, van Berkel TJ, Loskutoff
DJ, van Mourik JA. Tissue distribution of factor VIII gene expression
in vivo--a closer look. Thromb Haemost 2001; 86: 855-861. https://doi.org/10.1055/s-0037-1616143 PMid:11583319
- Madoiwa
S, Yamauchi T, Kobayashi E, Hakamata Y, Dokai M, Makino N, Kashiwakura
Y, Ishiwata A, Ohmori T, Mimuro J, Sakata Y. Induction of factor
VIII-specific unresponsiveness by intrathymic factor VIII injection in
murine hemophilia A. J Thromb Haemost 2009; 7: 811-824. https://doi.org/10.1111/j.1538-7836.2009.03314.x PMid:19220731
- Vremec
D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by
dendritic cell subtypes in mouse thymus and spleen. J Immunol 2000;
164: 2978-2986. https://doi.org/10.4049/jimmunol.164.6.2978 PMid:10706685
- Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol 2005; 5: 606-616. https://doi.org/10.1038/nri1669 PMid:16056254
- Navarrete
A, Dasgupta S, Delignat S, Caligiuri G, Christophe OD, Bayry J,
Nicoletti A, Kaveri SV, Lacroix-Desmazes S. Splenic marginal zone
antigen-presenting cells are critical for the primary allo-immune
response to therapeutic factor VIII in hemophilia A. J Thromb Haemost
2009; 7: 1816-1823. https://doi.org/10.1111/j.1538-7836.2009.03571.x PMid:19682235
- Qian
J, Burkly LC, Smith EP, Ferrant JL, Hoyer LW, Scott DW, Haudenschild
CC. Role of CD154 in the secondary immune response: the reduction of
pre-existing splenic germinal centers and anti-factor VIII inhibitor
titer. Eur J Immunol 2000; 30: 2548-2554. https://doi.org/10.1002/1521-4141(200009)30:9<2548::AID-IMMU2548>3.0.CO;2-H
- Feuerer
M, Beckhove P, Garbi N, Mahnke Y, Limmer A, Hommel M, Hammerling GJ,
Kyewski B, Hamann A, Umansky V, Schirrmacher V. Bone marrow as a
priming site for T-cell responses to blood-borne antigen. Nat Med 2003;
9: 1151-1157. https://doi.org/10.1038/nm914 PMid:12910264
- Stern
LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley
DC. Crystal structure of the human class II MHC protein HLA-DR1
complexed with an influenza virus peptide. Nature 1994; 368: 215-221. https://doi.org/10.1038/368215a0 PMid:8145819
- Celli
S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic
cell interactions in vivo reveals the importance of prolonged contacts
for CD4+ T cell activation. Immunity 2007; 27: 625-634. https://doi.org/10.1016/j.immuni.2007.08.018 PMid:17950004
- Lipscomb MF, Masten BJ. Dendritic cells: immune regulators in health and disease. Physiol Rev 2002; 82: 97-130. https://doi.org/10.1152/physrev.00023.2001 PMid:11773610
- Pfistershammer
K, Stockl J, Siekmann J, Turecek PL, Schwarz HP, Reipert BM.
Recombinant factor VIII and factor VIII-von Willebrand factor complex
do not present danger signals for human dendritic cells. Thromb Haemost
2006; 96: 309-316. https://doi.org/10.1160/TH05-11-0729 PMid:16953272
- Peerlinck
K, Arnout J, Gilles JG, Saint-Remy JM, Vermylen J. A higher than
expected incidence of factor VIII inhibitors in multitransfused
haemophilia A patients treated with an intermediate purity pasteurized
factor VIII concentrate. Thromb Haemost 1993; 69: 115-118. https://doi.org/10.1055/s-0038-1651565 PMid:8456422
- Peerlinck
K, Arnout J, Di GM, Gilles JG, Laub R, Jacquemin M, Saint-Remy JM,
Vermylen J. Factor VIII inhibitors in previously treated haemophilia A
patients with a double virus-inactivated plasma derived factor VIII
concentrate. Thromb Haemost 1997; 77: 80-86. https://doi.org/10.1055/s-0038-1655911 PMid:9031454
- Qadura
M, Waters B, Burnett E, Chegeni R, Bradshaw S, Hough C, Othman M,
Lillicrap D. Recombinant and plasma-derived factor VIII products induce
distinct splenic cytokine microenvironments in hemophilia A mice. Blood
2009; 114: 871-880. https://doi.org/10.1182/blood-2008-09-174649 PMid:19411636
- Bovenschen
N, Mertens K, Hu L, Havekes LM, van Vlijmen BJ. LDL receptor cooperates
with LDL receptor-related protein in regulating plasma levels of
coagulation factor VIII in vivo. Blood 2005; 106: 906-912. https://doi.org/10.1182/blood-2004-11-4230 PMid:15840700
- Lenting
PJ, Neels JG, van den Berg BM, Clijsters PP, Meijerman DW, Pannekoek H,
van Mourik JA, Mertens K, van Zonneveld AJ. The light chain of factor
VIII comprises a binding site for low density lipoprotein
receptor-related protein. J Biol Chem 1999; 274: 23734-23739. https://doi.org/10.1074/jbc.274.34.23734 PMid:10446132
- Bovenschen
N, Rijken DC, Havekes LM, van Vlijmen BJ, Mertens K. The B domain of
coagulation factor VIII interacts with the asialoglycoprotein receptor.
J Thromb Haemost 2005; 3: 1257-1265. https://doi.org/10.1111/j.1538-7836.2005.01389.x PMid:15946216
- Dasgupta
S, Navarrete AM, Bayry J, Delignat S, Wootla B, Andre S, Christophe O,
Nascimbeni M, Jacquemin M, Martinez-Pomares L, Geijtenbeek TB, Moris A,
Saint-Remy JM, Kazatchkine MD, Kaveri SV, Lacroix-Desmazes S. A role
for exposed mannosylations in presentation of human therapeutic
self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci U S A 2007;
104: 8965-8970. https://doi.org/10.1073/pnas.0702120104 PMid:17502612 PMCid:PMC1885611
- Dasgupta
S, Repesse Y, Bayry J, Navarrete AM, Wootla B, Delignat S, Irinopoulou
T, Kamate C, Saint-Remy JM, Jacquemin M, Lenting PJ, Borel-Derlon A,
Kaveri SV, Lacroix-Desmazes S. VWF protects FVIII from endocytosis by
dendritic cells and subsequent presentation to immune effectors. Blood
2007; 109: 610-612. https://doi.org/10.1182/blood-2006-05-022756 PMid:16985172
- Goudemand
J, Rothschild C, Demiguel V, Vinciguerrat C, Lambert T, Chambost H,
Borel-Derlon A, Claeyssens S, Laurian Y, Calvez T. Influence of the
type of factor VIII concentrate on the incidence of factor VIII
inhibitors in previously untreated patients with severe hemophilia A.
Blood 2006; 107: 46-51. https://doi.org/10.1182/blood-2005-04-1371 PMid:16166584
- Gouw
SC, van den Berg HM, le CS, van der Bom JG. Treatment characteristics
and the risk of inhibitor development: a multicenter cohort study among
previously untreated patients with severe hemophilia A. J Thromb
Haemost 2007; 5: 1383-1390. https://doi.org/10.1111/j.1538-7836.2007.02595.x PMid:17456190
- Delignat
S, Repesse Y, Navarrete AM, Meslier Y, Gupta N, Christophe OD, Kaveri
SV, Lacroix-Desmazes S. Immunoprotective effect of von Willebrand
factor towards therapeutic factor VIII in experimental haemophilia A.
Haemophilia 2012; 18: 248-254. https://doi.org/10.1111/j.1365-2516.2011.02679.x PMid:22044692
- Herczenik
E, van Haren SD, Wroblewska A, Kaijen P, van den Biggelaar M, Meijer
AB, Martinez-Pomares L, ten BA, Voorberg J. Uptake of blood coagulation
factor VIII by dendritic cells is mediated via its C1 domain. J Allergy
Clin Immunol 2012; 129: 501-509.e5. https://doi.org/10.1016/j.jaci.2011.08.029 PMid:21962992
- Meems
H, Meijer AB, Cullinan DB, Mertens K, Gilbert GE. Factor VIII C1 domain
residues Lys 2092 and Phe 2093 contribute to membrane binding and
cofactor activity. Blood 2009; 114: 3938-3946. https://doi.org/10.1182/blood-2009-01-197707 PMid:19687511
- Wroblewska
A, van Haren SD, Herczenik E, Kaijen P, Ruminska A, Jin SY, Zheng XL,
van den Biggelaar M, ten BA, Meijer AB, Voorberg J. Modification of an
exposed loop in the C1 domain reduces immune responses to factor VIII
in hemophilia A mice. Blood 2012; 119: 5294-5300. https://doi.org/10.1182/blood-2011-11-391680 PMid:22498747 PMCid:PMC3680040
- Bayry
J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Monoclonal antibody
and intravenous immunoglobulin therapy for rheumatic diseases:
rationale and mechanisms of action. Nat Clin Pract Rheumatol 2007; 3:
262-272. https://doi.org/10.1038/ncprheum0481 PMid:17471245
- Bayry
J, Siberil S, Triebel F, Tough DF, Kaveri SV. Rescuing CD4+CD25+
regulatory T-cell functions in rheumatoid arthritis by
cytokine-targeted monoclonal antibody therapy. Drug Discov Today 2007;
12: 548-552. https://doi.org/10.1016/j.drudis.2007.05.002 PMid:17631249
- Marcucci
M, Mancuso ME, Santagostino E, Kenet G, Elalfy M, Holzhauer S,
Bidlingmaier C, Escuriola EC, Iorio A, Nowak-Gottl U. Type and
intensity of FVIII exposure on inhibitor development in PUPs with
haemophilia A. A patient-level meta-analysis. Thromb Haemost 2015; 113:
958-967. https://doi.org/10.1160/TH14-07-0621 PMid:25631402
- Davis
SJ, Ikemizu S, Evans EJ, Fugger L, Bakker TR, van der Merwe PA. The
nature of molecular recognition by T cells. Nat Immunol 2003; 4:
217-224. https://doi.org/10.1038/ni0303-217 PMid:12605231
- Waters
B, Lillicrap D. The molecular mechanisms of immunomodulation and
tolerance induction to factor VIII. J Thromb Haemost 2009; 7:
1446-1456. https://doi.org/10.1111/j.1538-7836.2009.03538.x PMid:19583822
- Qian
J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor
VIII inhibitors in murine hemophilia A. Blood 2000; 95: 1324-1329. https://doi.org/10.1182/blood.V95.4.1324.004k25_1324_1329 PMid:10666206
- Reipert
BM, Sasgary M, Ahmad RU, Auer W, Turecek PL, Schwarz HP. Blockade of
CD40/CD40 ligand interactions prevents induction of factor VIII
inhibitors in hemophilic mice but does not induce lasting immune
tolerance. Thromb Haemost 2001; 86: 1345-1352. https://doi.org/10.1055/s-0037-1616733 PMid:11776297
- Rossi
G, Sarkar J, Scandella D. Long-term induction of immune tolerance after
blockade of CD40-CD40L interaction in a mouse model of hemophilia A.
Blood 2001; 97: 2750-2757. https://doi.org/10.1182/blood.V97.9.2750 PMid:11313267
- Ewenstein
BM, Hoots WK, Lusher JM, DiMichele D, White GC, Adelman B, Nadeau K.
Inhibition of CD40 ligand (CD154) in the treatment of factor VIII
inhibitors. Haematologica 2000; 85: 35-39.
- Kawai
T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic
complications after treatment with monoclonal antibody against CD40
ligand. Nat Med 2000; 6: 114. https://doi.org/10.1038/72162
- Koyama
I, Kawai T, Andrews D, Boskovic S, Nadazdin O, Wee SL, Sogawa H, Wu DL,
Smith RN, Colvin RB, Sachs DH, Cosimi AB. Thrombophilia associated with
anti-CD154 monoclonal antibody treatment and its prophylaxis in
nonhuman primates. Transplantation 2004; 77: 460-462. https://doi.org/10.1097/01.TP.0000110291.29370.C0 PMid:14966427
- LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood 2008; 112: 1570-1580. https://doi.org/10.1182/blood-2008-02-078071 PMid:18725575 PMCid:PMC2518873
- Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity 1998; 8: 363-372. https://doi.org/10.1016/S1074-7613(00)80541-5
- Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol 2005; 23: 367-386. https://doi.org/10.1146/annurev.immunol.23.021704.115723 PMid:15771575
- Hausl
C, Maier E, Schwarz HP, Ahmad RU, Turecek PL, Dorner F, Reipert BM.
Long-term persistence of anti-factor VIII antibody-secreting cells in
hemophilic mice after treatment with human factor VIII. Thromb Haemost
2002; 87: 840-845. https://doi.org/10.1055/s-0037-1613094 PMid:12038787
- Hausl
C, Ahmad RU, Sasgary M, Doering CB, Lollar P, Richter G, Schwarz HP,
Turecek PL, Reipert BM. High-dose factor VIII inhibits factor
VIII-specific memory B cells in hemophilia A with factor VIII
inhibitors. Blood 2005; 106: 3415-3422. https://doi.org/10.1182/blood-2005-03-1182 PMid:16091456 PMCid:PMC1895061
- Hausl
C, Ahmad RU, Schwarz HP, Muchitsch EM, Turecek PL, Dorner F, Reipert
BM. Preventing restimulation of memory B cells in hemophilia A: a
potential new strategy for the treatment of antibody-dependent immune
disorders. Blood 2004; 104: 115-122. https://doi.org/10.1182/blood-2003-07-2456 PMid:15001466
- Reipert
BM, Gangadharan B, Hofbauer CJ, Scheiflinger F, Bowen J, Donnachie E,
Fijvandraat K, Gruppo RA, Klintman J, Male C, McGuinn CE, Meeks SL,
Recht M, Ragni MV, Yaish HM, Santagostino E, Brown DL. Appearance of
high-affinity antibodies precedes clinical diagnosis of FVIII
inhibitors - Preliminary analysis from the Hemophilia Inhibitor PUP
Study (HIPS). Blood 128[22], 328. 2016. (Abstract) https://doi.org/10.1182/blood.V128.22.328.328
- Matino
D, Gargaro M, Santagostino E, Di Minno MN, Castaman G, Morfini M,
Rocino A, Mancuso ME, Di MG, Coppola A, Talesa VN, Volpi C, Vacca C,
Orabona C, Iannitti R, Mazzucconi MG, Santoro C, Tosti A, Chiappalupi
S, Sorci G, Tagariello G, Belvini D, Radossi P, Landolfi R, Fuchs D,
Boon L, Pirro M, Marchesini E, Grohmann U, Puccetti P, Iorio A,
Fallarino F. IDO1 suppresses inhibitor development in hemophilia A
treated with factor VIII. J Clin Invest 2015; 125: 3766-3781. https://doi.org/10.1172/JCI81859 PMid:26426076 PMCid:PMC4607121
- Palmer
DS, Dudani AK, Drouin J, Ganz PR. Identification of novel factor VIII
inhibitor epitopes using synthetic peptide arrays. Vox Sang 1997; 72:
148-161. https://doi.org/10.1159/000461983 PMid:9145485
- Huang
CC, Shen MC, Chen JY, Hung MH, Hsu TC, Lin SW. Epitope mapping of
factor VIII inhibitor antibodies of Chinese origin. Br J Haematol 2001;
113: 915-924. https://doi.org/10.1046/j.1365-2141.2001.02839.x PMid:11442484
- Lacroix-Desmazes
S, Bayry J, Misra N, Horn MP, Villard S, Pashov A, Stieltjes N, d'Oiron
R, Saint-Remy JM, Hoebeke J, Kazatchkine MD, Reinbolt J, Mohanty D,
Kaveri SV. The prevalence of proteolytic antibodies against factor VIII
in hemophilia A. N Engl J Med 2002; 346: 662-667. https://doi.org/10.1056/NEJMoa011979 PMid:11870243
- Scandella
D, Gilbert GE, Shima M, Nakai H, Eagleson C, Felch M, Prescott R,
Rajalakshmi KJ, Hoyer LW, Saenko E. Some factor VIII inhibitor
antibodies recognize a common epitope corresponding to C2 domain amino
acids 2248 through 2312, which overlap a phospholipid-binding site.
Blood 1995; 86: 1811-1819. https://doi.org/10.1182/blood.V86.5.1811.bloodjournal8651811 PMid:7544643
- Saenko
EL, Shima M, Rajalakshmi KJ, Scandella D. A role for the C2 domain of
factor VIII in binding to von Willebrand factor. J Biol Chem 1994; 269:
11601-11605.
- Zhong
D, Saenko EL, Shima M, Felch M, Scandella D. Some human inhibitor
antibodies interfere with factor VIII binding to factor IX. Blood 1998;
92: 136-142. https://doi.org/10.1182/blood.V92.1.136.413k35_136_142 PMid:9639509
- Lollar
P, Parker ET, Curtis JE, Helgerson SL, Hoyer LW, Scott ME, Scandella D.
Inhibition of human factor VIIIa by anti-A2 subunit antibodies. J Clin
Invest 1994; 93: 2497-2504. https://doi.org/10.1172/JCI117259 PMid:8200986 PMCid:PMC294465
- Lacroix-Desmazes
S, Moreau A, Sooryanarayana, Bonnemain C, Stieltjes N, Pashov A, Sultan
Y, Hoebeke J, Kazatchkine MD, Kaveri SV. Catalytic activity of
antibodies against factor VIII in patients with hemophilia A. Nat Med
1999; 5: 1044-1047. https://doi.org/10.1038/12483 PMid:10470082
- Lavigne-Lissalde
G, Lacroix-Desmazes S, Wootla B, Tarrade C, Schved JF, Kaveri SV,
Granier C, Villard-Saussine S. Molecular characterization of human B
domain-specific anti-factor VIII monoclonal antibodies generated in
transgenic mice. Thromb Haemost 2007; 98: 138-147. https://doi.org/10.1160/TH06-09-0510 PMid:17598006
- Klintman
J, Hillarp A, Berntorp E, Astermark J. Long-term anti-FVIII antibody
response in Bethesda-negative haemophilia A patients receiving
continuous replacement therapy. Br J Haematol 2013; 163: 385-392. https://doi.org/10.1111/bjh.12540 PMid:24032553
- Butenas S, Krudysz-Amblo J, Rivard GE, Mann G. Product-dependent anti-factor VIII antibodies. Haemophilia 2013; 19: 619-625. https://doi.org/10.1111/hae.12127 PMid:23557464 PMCid:PMC3688703
- Hofbauer
CJ, Whelan SF, Hirschler M, Allacher P, Horling FM, Lawo JP, Oldenburg
J, Tiede A, Male C, Windyga J, Greinacher A, Knobl PN, Schrenk G, Koehn
J, Scheiflinger F, Reipert BM. Affinity of FVIII-specific antibodies
reveals major differences between neutralizing and nonneutralizing
antibodies in humans. Blood 2015; 125: 1180-1188. https://doi.org/10.1182/blood-2014-09-598268 PMid:25515962
- Prescott
R, Nakai H, Saenko EL, Scharrer I, Nilsson IM, Humphries JE, Hurst D,
Bray G, Scandella D. The inhibitor antibody response is more complex in
hemophilia A patients than in most nonhemophiliacs with factor VIII
autoantibodies. Recombinate and Kogenate Study Groups. Blood 1997; 89:
3663-3671. https://doi.org/10.1182/blood.V89.10.3663 PMid:9160671
- Whelan
SF, Hofbauer CJ, Horling FM, Allacher P, Wolfsegger MJ, Oldenburg J,
Male C, Windyga J, Tiede A, Schwarz HP, Scheiflinger F, Reipert BM.
Distinct characteristics of antibody responses against factor VIII in
healthy individuals and in different cohorts of hemophilia A patients.
Blood 2013; 121: 1039-1048. https://doi.org/10.1182/blood-2012-07-444877 PMid:23243272
- White
GC, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune
responses in hemophilia: why do inhibitors develop in some, but not all
hemophiliacs? J Thromb Haemost 2005; 3: 1676-1681. https://doi.org/10.1111/j.1538-7836.2005.01375.x PMid:16102033
- Miao
CH, Ye P, Thompson AR, Rawlings DJ, Ochs HD. Immunomodulation of
transgene responses following naked DNA transfer of human factor VIII
into hemophilia A mice. Blood 2006; 108: 19-27. https://doi.org/10.1182/blood-2005-11-4532 PMid:16507778 PMCid:PMC1895820
- Peng
B, Ye P, Blazar BR, Freeman GJ, Rawlings DJ, Ochs HD, Miao CH.
Transient blockade of the inducible costimulator pathway generates
long-term tolerance to factor VIII after nonviral gene transfer into
hemophilia A mice. Blood 2008; 112: 1662-1672. https://doi.org/10.1182/blood-2008-01-128413 PMid:18574023 PMCid:PMC2518877
- Bray
GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, Ragni MV,
Goedert JJ. Loss of high-responder inhibitors in patients with severe
hemophilia A and human immunodeficiency virus type 1 infection: a
report from the Multi-Center Hemophilia Cohort Study. Am J Hematol
1993; 42: 375-379. https://doi.org/10.1002/ajh.2830420408 PMid:8493988
- Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301-305. https://doi.org/10.1126/science.1071059 PMid:11951032
- Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol 2012; 3: 287. https://doi.org/10.3389/fimmu.2012.00287 PMid:23060876 PMCid:PMC3443751
- Pordes
AG, Baumgartner CK, Allacher P, Ahmad RU, Weiller M, Schiviz AN,
Schwarz HP, Reipert BM. T cell-independent restimulation of
FVIII-specific murine memory B cells is facilitated by dendritic cells
together with toll-like receptor 7 agonist. Blood 2011; 118: 3154-3162.
https://doi.org/10.1182/blood-2011-02-336198 PMid:21788339
- Reding MT. Immunological aspects of inhibitor development. Haemophilia 2006; 12 Suppl 6: 30-35. https://doi.org/10.1111/j.1365-2516.2006.01363.x PMid:17123391
- Reding
MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1-
and Th2-induced anti-factor VIII IgG subclasses in congenital and
acquired hemophilia patients. Thromb Haemost 2002; 88: 568-575. https://doi.org/10.1055/s-0037-1613257 PMid:12362225
- Reding
MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+
T-cell epitope repertoire on the C2 domain of coagulation factor VIII.
J Thromb Haemost 2003; 1: 1777-1784. https://doi.org/10.1046/j.1538-7836.2003.00251.x PMid:12911593
- Reding
MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope
repertoire of human CD4(+) T cells on the A3 domain of coagulation
factor VIII. J Thromb Haemost 2004; 2: 1385-1394. https://doi.org/10.1111/j.1538-7836.2004.00850.x PMid:15304045
- Reipert
BM, van Helden PM, Schwarz HP, Hausl C. Mechanisms of action of immune
tolerance induction against factor VIII in patients with congenital
haemophilia A and factor VIII inhibitors. Br J Haematol 2007; 136:
12-25. https://doi.org/10.1111/j.1365-2141.2006.06359.x PMid:17222196
- Cao
O, Loduca PA, Herzog RW. Role of regulatory T cells in tolerance to
coagulation factors. J Thromb Haemost 2009; 7 Suppl 1: 88-91. https://doi.org/10.1111/j.1538-7836.2009.03417.x PMid:19630776 PMCid:PMC2911015
- Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 2008; 9: 239-244. https://doi.org/10.1038/ni1572 PMid:18285775 PMCid:PMC3075612
- Key NS. Inhibitors in congenital coagulation disorders. Br J Haematol 2004; 127: 379-391. https://doi.org/10.1111/j.1365-2141.2004.05168.x PMid:15521914