Samir K. Ballas.
Cardeza Foundation for
Hematologic Research, Department of Medicine, Sidney Kimmel Medical
College, Thomas Jefferson University, Philadelphia, PA, USA.
Corresponding
author: Samir K. Ballas MD FACP. Cardeza Foundation, Department of
Medicine, Sidney Kimmel Medical College, Thomas Jefferson University,
1020 Locust Street, Philadelphia, PA 19107. Tel: 856 745 6380, Fax: 856
795 0809. E-mail:
samir.ballas@jefferson.edu
Published: January 1, 2020
Received: November 25, 2019
Accepted: December 17, 2019
Mediterr J Hematol Infect Dis 2020, 12(1): e2020010 DOI
10.4084/MJHID.2020.010
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Sickle
cell disease (SCD) is an extremely heterogeneous disease that has been
associated with global morbidity and early mortality. More effective
and inexpensive therapies are needed. During the last five years, the
landscape of the pharmacotherapy of SCD has changed dramatically.
Currently, 54 drugs have been used or under consideration to use for
the treatment of SCD. These fall into 3 categories: the first category
includes the four drugs (Hydroxyurea, L-Glutamine, Crizanlizumab tmca
and Voxelotor) that have been approved by the United States Food and
Drug Administration (FDA) based on successful clinical trials. The
second category includes 22 drugs that failed, discontinued or
terminated for now and the third category includes 28 drugs that are
actively being considered for the treatment of SCD. Crizanlizumab and
Voxelotor are included in the first and third categories because they
have been used in more than one trial. New therapies targeting multiple
pathways in the complex pathophysiology of SCD have been achieved or
are under continued investigation. The emerging trend seems to be the
use of multimodal drugs (i.e. drugs that have different mechanisms of
action) to treat SCD similar to the use of multiple chemotherapeutic
agents to treat cancer.
|
Introduction
Sickle
cell anemia (SCA) is among the most common inherited hemolytic anemias,
and affects an estimated 100,000 persons in the US and probably
millions worldwide.[1] The true global incidence of
sickle cell disease (SCD) is unknown. The World Health Organization has
estimated that each year 220,000 babies are born with SCD in Africa,
and that SCD accounts for up to 16% of deaths of children aged < 5
years in some African countries.[2,3] The reported
prevalence of the sickle cell trait in African Americans varies from
6.7 to 10.1% and in Africans the range is from 10 to 40% across
equatorial Africa and decreases to between 1 and 2% on the North
African coast and < 1% in South Africa.[4-6] The prevalence of the sickle cell trait varies widely worldwide and may be as high as 50% in certain regions.[6-8] The prevalence of SCA is ~ 1 in 600 newborn African American infants and 150,000 - 300,000 newborn Africans.[9-11]
Sickle
cell anemia is a hereditary disorder of hemoglobin (Hb) where the
sickle gene is inherited, homozygously, from both parents. The sickle
mutation is the result of a single base change (GAG → GTG) in the sixth
codon of exon 1 of the β-globin gene responsible for the synthesis of
the β-globin polypeptide of the Hb molecule (α2β2). This change, in
turn, results in replacement of a normal glutamic acid with valine at
position 6 of the β-globin chain and the formation of sickle Hb. Sickle
erythrocytes are rigid with decreased deformability and reduced life
span resulting in hemolysis, vaso-occlusive disease, vasculopathy and
subsequent inflammation and end organ damage.[12,13]
Clinical
manifestations of SCD include pain syndromes, anemia and its sequelae,
organ failure including infection/inflammation and comorbid conditions.[14]
The painful acute vaso-occlusive crisis (VOC) is the hallmark of SCD
and traditionally, has been thought to be to be due to sickle
erythrocytes occluding the microvasculature, especially within bones,
and causing tissue ischemia, injury, and pain. Recent studies, however,
suggest that the mechanism is a more complex process that is
multicellular, involving interactions with the vascular endothelium, as
well as contributions from hemolysis, inflammation, and coagulation.[15]
Despite having a common genetic basis and similar pathophysiology,
individual patients with SCA have a highly variable clinical phenotype.
The prevalence of these complications varies with age from infancy
through adult life as shown in Figure 1. However, pain, infections and anemia requiring blood transfusion occur throughout the life span of affected patients.
Clinical
care for affected individuals has been mostly palliative, including
supportive, symptomatic, preventative and abortive approaches, as shown
in Table 1.
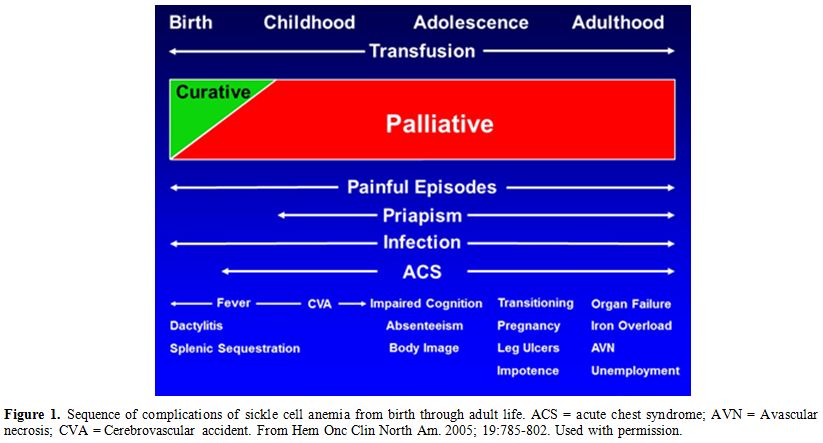 |
Figure 1.
Sequence of complications of sickle cell anemia from birth
through adult life. ACS = acute chest syndrome; AVN = Avascular
necrosis; CVA = Cerebrovascular accident. From Hem Onc Clin North Am.
2005; 19:785-802. Used with permission. |
 |
Table 1. Palliative Management of Sickle Cell Disease and its Complications. |
Advances
in the management of SCD beyond palliation include pharmacotherapy and
curative cellular therapies. The latter include stem cell
transplantation and gene therapy[15,16] and these
will not be addressed in this review. In addition, some of the current
approaches to the management of SCD could be pharmacologic or
nonpharmacologic, especially when it comes to pain management. Examples
of nonpharmacologic treatments include meditation, therapeutic massage,
transcutaneous electrical nerve stimulation, heat and cold packs,
distraction, relaxation, music, guided imagery, self-hypnosis,
acupuncture and biofeedback.[13,17]
Current examples of pharmacologic therapies include the use of
non-steroidal anti-inflammatory drugs, opioids, adjuvants, steroids,
and so on.[13] The aim of this study is to review the
current status of pharmacotherapy for the treatment of SCD,
Historically, pharmacotherapeutic drugs that have been tried to treat
SCD fall into three groups. The first group includes the successful
drugs approved by the FDA shown in Table 2. The second group includes the drugs that were tried but failed to show a beneficial effect shown in Table 3. The third group includes potential drugs that are being used in different phases of randomized clinical trials shown in Table 4 and will be discussed below.
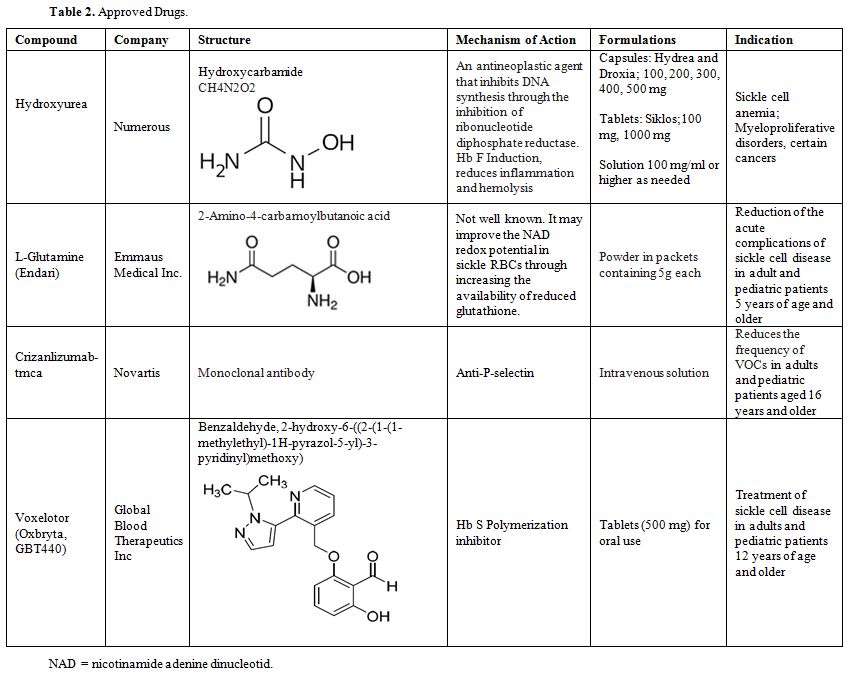 |
Table 2. Approved Drugs. |
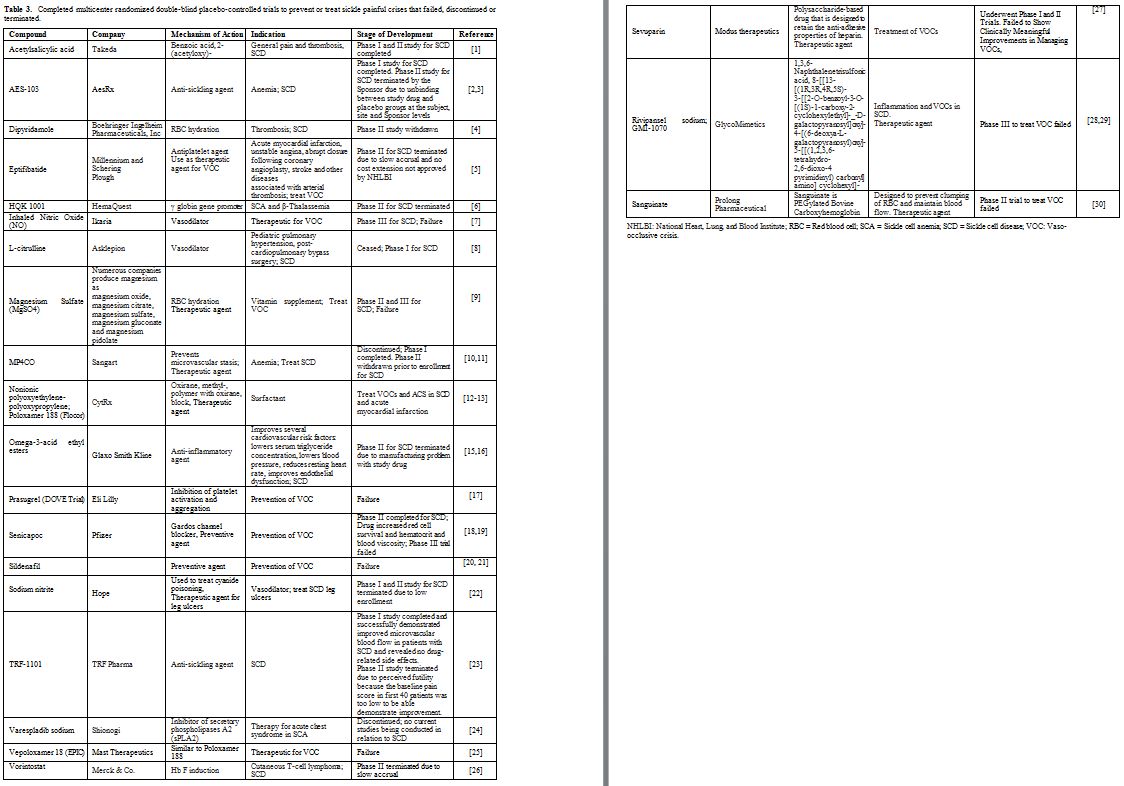 |
Table 3. Completed multicenter randomized
double-blind placebo-controlled trials to prevent or treat sickle
painful crises that failed, discontinued or terminated. |
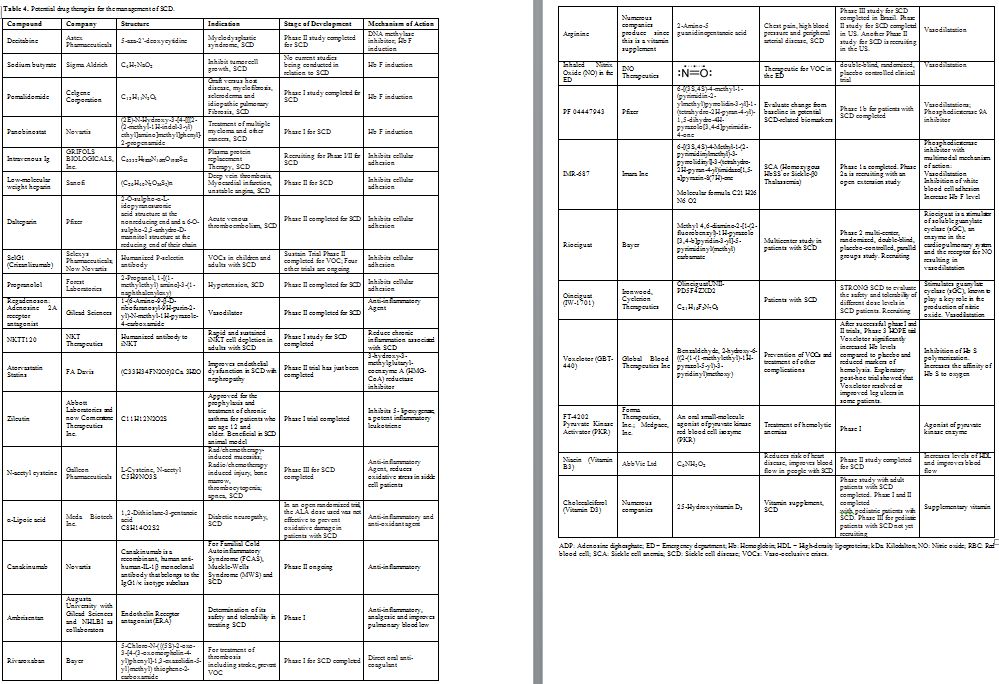 |
Table 4. Potential drug therapies for the management of SCD. |
The Economic Burdens of SCD
Sickle
cell disease is a global disease affecting millions of people worldwide
and hundreds of thousands in the US. It affects not only those of
African descent, but also persons of Middle Eastern, Indian, Latin
American and Mediterranean descent. It has received very little
attention and even less research funding. National Institute of Health
(NIH) grants for sickle cell research were much less than that for
less-common inherited diseases. In 1972, the National Sickle Cell
Anemia Control Act was signed, which paved the way for more research
funding and established screening and education programs. The NIH
dedicated $10 million to be spent on SCD research at that time.[13] The economic burden to patients with SCD is significant.[18-22]
Many patients are living in poverty with their illness due to chronic
pain, and physical disability limiting their ability to work and
contribute to society.[13] The economic burden on society was estimated at $1.1 billion in 2009.[18]
This number is projected to increase as patients with SCD are living
longer as we continue to improve supportive care. A solution to this
problem is not simple, requiring multidisciplinary action with
increased funding, legislation, research and supportive services.
Simple therapy with hydroxyurea (HU) is still not available to the
millions in Africa today. As we continue to push for new therapies for
SCD, HU continues to have tremendous potential in the global
marketplace.
Evolution of the Approaches to Treat SCD
Since
sickled cells were first described in 1910 and the mutation causing
abnormal Hb S was identified in 1949, the complex mechanism underlying
its pathophysiology continues to evolve.[23] A
cascade of events driven by endothelial damage and inflammation leads
to vasculopathy. The inciting event is injury to the red blood cell
(RBC) membrane. Hemoglobin S polymerization impairs deformability of
the RBC and causes oxidative injury and destruction of the RBC. RBC
injury exposes phosphatidyl serine and releases Hb and other
intracellular contents. This in turn depletes NO, increases endothelial
adherence, releases proinflammatory cytokines and activates the
coagulation cascade causing ischemia, reperfusion injury and vascular
damage.[12,17,23]
Damaged
sickle cells are prone to adhere to the endothelium by adhesion
molecules. The RBC membrane receptors VLA-4/a4b1 bind to endothelial
receptors directly to vascular cell adhesion molecule 1 (VCAM-1) and
interacts with subendothelial matrix proteins (BCAM/LU, a4b1 with the
laminin and von Willebrand factor).[24,25] Red blood
cell interactions with the vascular endothelium also lead to the
production of oxygen radicals by activating transcription factor
nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB).
NF-kB upregulates the production of endothelial adhesion molecules such
as E-selectin, VCAM-1 and intracellular adhesion molecule-1 (ICAM-1).
P-selectin and E-selectin on endothelial cells have been suggested to
participate in.[26,27]
In preclinical studies an
anti-P-selectin molecule showed increased microvascular flow and
reduced adhesion of leukocytes to the endothelium.[26]
ICAM-4, another RBC membrane protein, which participates in adhesion,
can be activated by epinephrine to adhere to endothelial membrane and
exacerbate vaso-occlusive disease and also increased leukocyte adhesion
to endothelium.[27] When treated with propranolol (a b-adrenergic receptor antagonist) VOCs were diminished.[28,29]
In
addition to adherence to endothelial cells, RBCs in SCA also adhered
strongly to leukocytes in VOCs via interactions with P-selectin and E
selectin. This interaction is propagated by TNF-a. Selectins function
in adhesion to the vessel wall by recruiting rolling particles and
cells and also contribute to cell activation. Patients with SCD have
chronic elevation of proinflammatory cytokines at baseline, including
C-reactive protein, TNF, IL-1 and IL-8. Damaged RBCs, activated
endothelial cells, leukocytes and platelets (PLTs) contribute to a
proinflammatory environment. Sickled RBCs stimulate endothelial cells
to release TNF-α and IL-1β. There is increased production of placental
growth factor, which activates monocytes to release reactive oxygen
species (ROS), which enhances inflammation.
Additionally,
invariant natural killer T (iNKT) cells are activated in patients with
SCD, suggesting that iNKT cells may play a critical role in mediating
inflammation. Intravascular hemolysis results in release of cell-free
Hb in plasma, and hemin release that contribute to the inflammation.[25,30]
Nitric oxide (NO) is produced by the endothelium from arginine and
causes vasodilation by binding to endothelin-1, a vasoconstrictor.
Intravascular hemolysis releases Hb, which scavenges NO in the plasma
and subendothelial spaces.
Depletion of NO leads to
vasoconstriction and formation of ROS. Nitric oxide also downregulates
adhesion molecules, VCAM-1, ICAM-1 and E-selectin. Erythrocyte arginase
released during hemolysis decreases arginine levels and decreases NO
production. The byproducts of these reactions, urea, proline,
polyamines and free radicals, cause vascular remodeling and
vasculopathy. Patients with SCD have elevated asymmetric
dimethylarginine, which inhibits arginine transport and promotes
endothelial dysfunction.[17,31,32]
These
inflammatory processes activate the coagulation cascade.
Phosphatidylserine expression on RBC surface and microparticles
activates tissue factor and, in turn, the extrinsic coagulation
cascade. Tissue factor also promotes inflammation and endothelial
damage. In preclinical studies in transgenic sickle mice, lowering
tissue factor levels resulted in lower plasma levels of IL-6 and
soluble VCAM-1.[33] Sickle cell disease is a chronic
inflammatory state and ROS are increased at baseline compared with
normal controls. Hemolysis releases Hb, and iron products, which
increase ROS that generate superoxide (O2-) and peroxynitrate (ONOO-),
which promotes an inflammatory response and causes cell death. Patients
with SCD have impaired buffer system with decreased glutathione, and
other antioxidants.[34-36]
Approved Pharmacotherapeutic Drugs
The
ideal drug for SCD would have analgesic properties, be able to prevent
VOCs or abort them with a rapid onset of action, would decrease the
severity and frequency of VOCs, have limited hazardous side-effect
profile and be effective in all patients, and available globally.
Currently HU, L-glutamine, Crizanlizumab tmca and Voxelotor shown in Table 2, are the only agents that fit some of these criteria and are approved by the FDA.
Hydroxyurea.
Hydroxyurea has many qualities of the ideal drug for SCD. It was
first synthesized in 1869 and used in myeloproliferative disorders.
Chemically it is a synthetic urea analog; also referred to as
hydroxycarbamide (HC) that functions as an antineoplastic agent. In
this review HU and HC are used synonymously. There is seemingly a
tendency to use the HU acronym in the US and HC acronym in the
UK. Hydroxyurea was identified as a potent Hb F inducer and was
subsequently found to be both a feasible and effective treatment option
for SCA.[13] It decreases the frequency of VOCs,
acute chest syndrome (ACS), and the frequency of blood transfusion. In
addition, HU improves the quality of life and decreases mortality in
patients with SCA.[37] However, HU is not effective in about 25% of those with SCA, an acronym that also includes sickle-β0-thalassemia (S-β0-T).[38] Currently, it was found to be teratogenic and possibly carcinogenic in animal studies[39]
but not in humans so far. It was the first pharmacotherapeutic drug to
be approved by the FDA and by the European Medicines Agency (EMA) for
the treatment of SCA.
Hydroxyurea is cell cycle specific for the S
phase and inhibits DNA synthesis as a ribonucleotide reductase
inhibitor. It induces the production of Hb F in the majority of
patients with SCA who are compliant with therapy and thus prevents the
formation of Hb S polymers.
The molecular mechanisms by which HU
induces Hb F production are not fully clear. Proposed mechanisms
include selectively killing cells in the bone marrow, and increasing
the number of early erythroid progenitors such as fetal erythroblasts
that lead to production of Hb F. It also reduces the number of adhesive
reticulocytes[40] and circulating inflammatory cells
such as monocytes and neutrophils. It alters circulating monocyte
subsets and dampens the inflammatory potential of SCD.[41,42] It also improves RBC deformability.[43] More recently, HU was reported to have antioxidant activity.[44]
It appears that patients whose high neutrophil and reticulocyte counts
decrease significantly after HU therapy have a higher increase in Hb F
levels.[3,21,45]
In addition, HC affects the plasma proteome of children with SCA
resulting in reduced inflammation and decreased activation of the
coagulation factors.[46] The increased Hb F induced
by HU decreases the biomarkers of oxidative stress and the scavenging
of NO in both sickle cell mice and in patients with SCD. [44,47,48]
More
complex effects of HU involve the production of NO, guanylyl cyclase
and cGMP dependent protein kinase pathway important in inducing
expression of the γ-globin gene. Additionally, HU improves erythrocyte
deformability, lowering of circulating leukocytes and reticulocytes,
and reduces hemolysis.[3,49,50] Since its first clinical application reported in 1984 by Platt et al., many trials were performed.[51]
The Multicenter Study of HU in SCA, a placebo-controlled randomized
Phase III trial of 299 adults with severe SCA, terminated early due to
significant reductions in frequency of VOC, ACS, need for blood
transfusion and delayed onset of first VOC.[52,53]
This study led to the FDA approval of HU for therapy on February 25,
1998 for moderately or severely affected adults with SCA. The Pediatric
Hydroxyurea Phase III Clinical Trial (BABY HUG), involving infants with
SCA randomized either to HU (fixed dose 20 mg/kg/day) or placebo. This
trial showed that HU did not clearly prevent organ damage, the primary
endpoint of the 2-year treatment period, but significantly decreased
the secondary endpoints: pain, ACS, hospitalizations, and transfusions
in children.[54-59]
Formulations of HU are shown in Table 2. It is available as capsules or tablets. Solutions of 100 mg/ml or higher can be prepared by pharmacist as needed.[60]
The usual staring dose is 15 mg/k/day. This may be increased gradually
every month as needed to achieve the maximum tolerable dose. Some
providers maintain a dose that increases Hb F to a desirable level
before achieving the maximum tolerable dose.
The common side effects of HU are listed in Table 5.
Toxic effects are dose and time dependent and can be prevented by
careful monitoring and surveillance. Side effects are generally
reversible with cessation or decrease of the drug dose. Hydroxyurea is
myelosuppressive and leukopenia is the most common manifestation
followed by thrombocytopenia and anemia. Macrocytosis is common and may
mask folic acid deficiency, so folic acid supplementation is
recommended during treatment with HU. Idiosyncratic side effects are
rare, reversible and more common in generic formulations.[61] Figure 2 shows an example of HU-induced melanonychia.
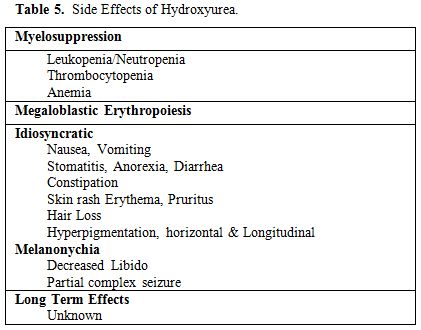 |
Table 5. Side Effects of Hydroxyurea. |
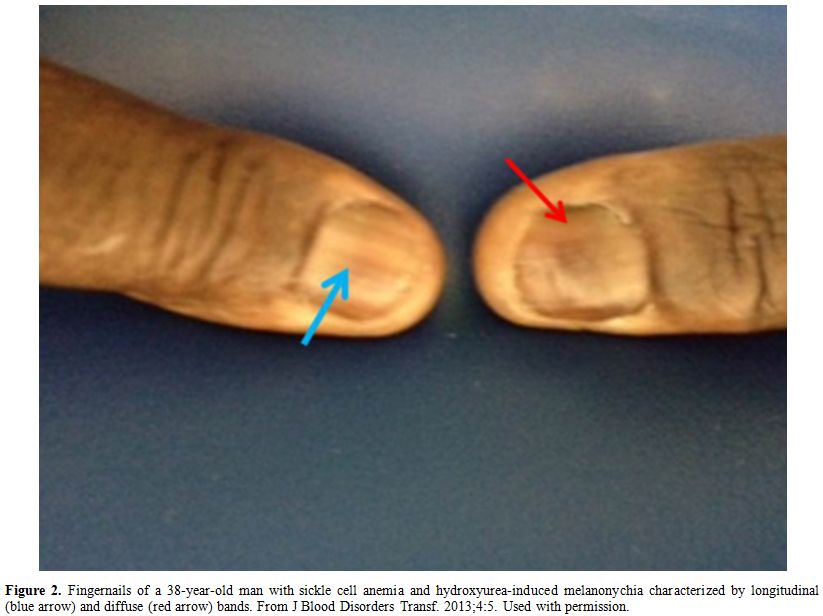 |
Figure 2. Fingernails of a 38-year-old man
with sickle cell anemia and hydroxyurea-induced melanonychia
characterized by longitudinal (blue arrow) and diffuse (red arrow)
bands. From J Blood Disorders Transf. 2013;4:5. Used with permission. |
Phase
IV of the HU study which refers to its use in the general population
post-approval by the FDA, showed a plethora of publications globally
addressing various aspects of its pros and cons. Most important among
these are as described below.
a. Adherence to HU Therapy.
The BABY HUG trial, which demonstrated safety and efficacy of starting
HU in infancy contributed to a robust increase in HU prescribing for
children with SCD.[62] Hydroxyurea use in infants 5-12 months old resulted in a better response compared with use in older patients.[63]
Moreover, prospective longitudinal follow-up of children with SCD
treated with HU since infancy was highly effective in preventing
complications of SCD.[64] Pediatric hematologists strongly recommend the use of HU in children with SCD early and frequently.[65]
Unfortunately,
access to specialist care for adolescents and adults with SCD is
limited and associated with many barriers. Most important among these
include appointment non-adherence.[66] Factors that
seem to influence these barriers may be provider- or patient-related.
Thus, patients who felt their providers were not listening to their
concerns tended to be non-adherent to HU therapy.[67]
Similarly,
at the global level the use of HU for the treatment of patients with
SCD varied considerably. The universal administration of HU to children
with SCD was successful in Malawi[68] but not in Nigeria [69]
where concerns about its long-term safety and toxicity limited its
prescription by physicians and acceptability by patients. The major
barriers to the use of HU in the treatment of SCD in Nigeria included
lack of national guidelines for the use of HU, concerns for infertility
and safety profile of HU in pregnancy and lactation.[69]
b. Hydroxyurea and Stroke.
According to the Cooperative Study of SCD (CSSCD), stroke occurred in
11% of children with SCA younger than 20 years of age and 24% of adults
by the age 45.[70] However, the use of transcranial
Doppler (TCD) in the Stroke Prevention in SCA (STOP 1) trial to
identify persons at higher risk for ischemic stroke, along with the
prophylactic management of those patients with chronic transfusion
(simple or RBC exchange), has dramatically reduced the incidence of
childhood primary stroke to 2% to 3%.[71,72] The STOP
2 trial determined that regular transfusion for primary stroke
prevention could not be halted safely, even in patients with a normal
magnetic resonance angiogram whose TCD results have normalized.[72,73]
Discontinuation of transfusions after 30 months resulted in a high rate of reversion to abnormal TCD velocity and stroke.[72,73]
A number of studies indicate that transfusion to prevent the recurrence
of strokes should be performed indefinitely, even after transition to
adult programs.[74-76] The advent of HU raised the
possibility if it could replace or decrease the need for transfusion to
prevent the recurrence of stroke. However, the Stroke with transfusions
changing to HU (SWITCH) trial and the Transcranial doppler with
transfusions changing to HU (TWITCH) trial were not successful [77,78]
and blood transfusion and iron chelation therapy remain the better
choice for the prevention of primary and secondary stroke in patients
with SCA. Nevertheless, HU treatment of children with SCA is associated
with more intact brain white matter integrity by using quantitative MRI[79] and prevents the conversion to abnormal transcranial doppler in SCA.[80]
The NIH guidelines for the management of SCD indicated that if it is
not possible to implement a transfusion program in children and adults
who have had a stroke, then HU therapy is recommended.[38]
c. Hydroxyurea and Leg ulcers.
The effect of HU on leg ulcers in patients with SCD is controversial,
though it has been reported to cause leg ulcers in patients with
myeloproliferative syndromes.[81] Data on leg
ulcers from the Cooperative Study of Sickle Cell Disease (CSSCD)
identified five risk factors associated with leg ulcers in patients
with SCD.[82] Leg ulcers were more common in males
and older patients and less common in patients with α-gene deletion,
high total Hb level and high levels of Hb F. Since HU is known to
increase total Hb level and Hb F, one would expect that HU would be
protective against the development of leg ulcers. Nevertheless, there
are anecdotes of leg ulcers occurring after therapy with HU and of
healed old ulcers reactivated after HU therapy.[83]
de Montalembert et al followed a cohort of 101 children with SCD
treated with HU for a median of 22 months; among these only one 18
year-old patient had leg ulcers 23 months after treatment.[84]
d. Hydroxyurea: pregnancy and lactation.
The FDA developed a system to rate medications and drugs based on
potential benefits and risks to the fetus. Drugs are classified into
pregnancy categories A, B, C, D, and X where A is safe and X
contraindicated. Hydroxyurea is classified as a category D drug; these
drugs have positive evidence human fetal risk but use may be justified
in some circumstances. Because HU, an S-phase antineoplastic drug, is
known to be carcinogenic, mutagenic, and teratogenic in animals, a
major inclusion criterion in the Multicenter Study of HU in SCA (MSH)
was the use of contraceptives both by females and males, to avoid fetal
exposure to HU. Despite this precautionary measure, some women have
become pregnant while they or their male partners were taking HU.
Surviving patients enrolled in the original MSH trial for up to 17
years post randomization were followed.[37] The
findings suggested that exposure of the fetus to HU did not cause
teratogenic changes in those pregnancies that terminated in live birth,
whether full term or premature.[39] This appears to
be true whether the parent taking HU was the mother or the father.
Safety of HU during pregnancy and SCD was also reported in 3 other
patients.[85,86] Safety of HU during pregnancy was also reported in other hematologic disorders.[86]
The
NHLBI evidence-based SCD guidelines identified the safety of HU during
gestation and subsequent lactation as an important knowledge gap that
requires further investigation. A clinical trial for that purpose is
underway.[87]
Similarly, breastfeeding is
usually contraindicated during maternal therapy with
antineoplastic drugs, but the evidence of this recommendation for HU is
very weak.[38,88] Current recommendations state that breastfeeding should be avoided for at least 3 hours after the mother takes HU.[89]
Currently, clinical trial [NCT02990598]: Hydroxyurea Exposure in
Lactation A Pharmacokinetics Study (HELPS) (HELPS) is underway to
examine the pharmacokinetics and distribution of oral HU when
administered as a single dose to lactating women.[90]
L-Glutamine (Endari). L-glutamine is an amino acid used in the synthesis of protein. It is the most abundant amino acid in human blood.[91]
The body can usually synthesize sufficient amounts of L-glutamine, but
in some instances of stress, the body's demand for glutamine increases,
and glutamine must be obtained from the diet. Accordingly, it is a
non-essential and conditionally essential amino acid in humans. Reduced
glutathione is the primary buffer for reactive oxygen species (ROS).
L-glutamine
is metabolized to glutamate, the glutathione precursor, and preserves
intracellular nicotinamide adenine dinucleotide (NAD), which is
necessary for glutathione recycling. Oral supplementation of glutamine
in SCD increases the NAD redox potential and may reduce sickle
erythrocyte adhesiveness.[32,33] Decreased NAD redox
potential due to low level of L-glutamine was a major mechanism for the
presence sickle RBCs under oxidant stress conditions.[92]
Oral glutamine was developed by Emmaus Medical for the treatment of
short bowel syndrome and in SCA and β thalassemia. It decreases the
resting energy expenditure in children with SCD. A multicenter Phase
III trial of L-glutamine supplementation in 230 children to prevent VOC
is completed; results wed that the median number of pain crises over 48
weeks was lower among those who received oral therapy with L-glutamine,
administered alone or with HU, than among those who received placebo,
with or without HU.[92-95] Two Phase II trials are also completed.[96,97]
Endari
was approved by the FDA on July 7, 2017 to reduce the acute
complications of SCD in adult and pediatric patients 5 years of age and
older.[98] It is available as an oral powder: 5 grams
of L–glutamine as a white crystalline powder in paper-foil-plastic
laminate packets. It should be administered orally, twice per day at
the dose based on body weight as follows: 5 g twice daily for patients
weighing < 30 Kg, 10 g twice daily for patients weighing 30-65 Kg
and 15 g twice daily for patients weighing > 65 kg. Side effects of
Endari included low-grade nausea, noncardiac chest pain, fatigue, and
musculoskeletal pain occurred more frequently in the l-glutamine group
than in the placebo group. There are no available data on Endari use
during pregnancy and lactation.
The efficacy of L-Glutamine in the
management of SCD awaits the data generated in phase IV post approval
in the general population of patients with SCD.
Crizanlizumab tmca (ADAKVEO).
The efficacy of SelG1 (Crizanlizumab), a humanized anti-P-selectin
monoclonal antibody, in preventing VOCs was evaluated in Phase II
SUSTAIN trial in combination with or without HU.[99]
Crizanlizumab intravenous therapy resulted in a significantly lower
rate of sickle cell-related VOCs than placebo and was associated with a
low incidence of adverse events.[99] The FDA approved
crizanlizumab-tmca (ADAKVEO, Novartis) on November 15, 2019 to reduce
the frequency of VOCs in adults and pediatric patients aged 16 years
and older with SCD.[100] The recommended dose is 5
mg/kg intravenously over a period of 30 minutes on week 0, 2, and every
4 weeks thereafter. The most common side effects (>10%) were nausea,
arthralgia, back pain, and pyrexia.
Voxelotor (Oxbryta, GBT440).
Voxelotor is an inhibitor of Hb S polymerization indicated for the
treatment of SCD in adults and children 12 years of age and older.
It exerts its action by biding to the amino acid terminal of both α
chains of Hb. The efficacy and safety of Voxelotor (OXBRYTA) in SCD was
evaluated in a Phase III randomized, double-blind, placebo-controlled
multicenter trial in combination with and without HU (HOPE Trial).[101,102] It was approved by the US FDA on November 19, 2019.[103]
The approval was accelerated based on increase in Hb. Continued
approval for this indication may be contingent upon verification and
description of clinical benefit in confirmatory trial(s).
Efficacy
was based on Hb response rate defined as a Hb increase of >1 g/dL
from baseline to Week 24 in patients treated with OXBRYTA 1,500 mg
versus placebo. The response rate for OXBRYTA 1,500 mg was 51.1%
(46/90) compared to 6.5% (6/92) in the placebo group (p < 0.001).
Recommended
dosage of OXBRYTA is 1,500 mg orally once daily with or without
food. Recommended dosage for severe hepatic impairment is 1,000
mg orally once daily with or without food. The daily dose of OXBRYTA
has to be adjusted in the presence of concomitant medications. Thus, in
the presence of strong CYP3A4 inhibitors or fluconazole, the dose
should be decreased to 1000 mg once daily. On the other hand, in the
presence of strong or moderate CVP3A4 inducers the recommended dose
should be increased to 2,500 mg once daily.[103]
Pending Pharmacotherapeutic Drugs for the Treatment of SCD
Currently,
there are at least 50 unapproved pharmacotherapeutic drugs that were or
are being used or tried to treat SCD during the last two decades. Most
of these were multicenter randomized double-blind placebo-controlled
trials to prevent or treat sickle painful VOCs. Preventive
pharmacotherapy includes drugs that are taken routinely as outpatients
with the hope that may decrease the frequency of VOCs that require
treatment in the emergency department or hospital. Therapeutic
pharmacotherapy includes drugs that are administered after admission to
the hospital with the hope that they may abort the VOC and decrease the
length of hospital stay and the amount of analgesics used. Twenty-two
of these drugs, shown in Table 3, failed, discontinued or terminated.
Among the 22 drugs listed in Table 3,
Rivipansel sodium (GMI-1070), has an interesting history that
demonstrates the steps a drug has to go through in order to achieve
approval. It is a small-molecule pan-selectin inhibitor that binds to
E, P and L selectin that was developed by Glycomimetic to target
inflammation in sickle VOCs. It improves blood flow by inhibiting
E-selectin and neutrophil activation. A randomized, double-blind,
placebo-controlled Phase II trial in 76 subjects hospitalized for
sickle cell VOC assessing GMI-1070 is complete. Data showed that the
patients treated with rivipansel sodium experienced reduction in
duration of VOC, length of hospital stay and reduction in the use of
opioids for pain relief. Both adult and pediatric patients demonstrated
improvement and adverse event rates were comparable between rivipansel
sodium and placebo.[104,105] However, Phase III of the study failed.
Failure of the 22 drugs listed in Table 3
teaches us at least two important lessons. First, most of the drugs
that went through phase III trials failed to treat or abort VOCs or
ACS. The approved drugs prevented or decreased the frequency of VOCs.
The second lesson is that hydration of sickle RBC does not seem to be
an adequate approach in the management of SCD. In the last 2-3 decades
hydration of sickle RBC was one of the major approaches to treat SCD.
The phase III Senicapoc trial showed that hydration of sickle
erythrocytes is counterproductive. This study concluded that hydration
of sickle RBC improves their survival which, in turn, increases the
blood hematocrit. Consequently, higher hematocrit is associated with
increased blood viscosity that promotes vaso-occlusion and the
precipitation of a new VOC.
The remaining 28 drugs that are not
approved by the FDA so far but are being used in different stages of
clinical trials to prevent or treat VOCs are listed in Table 4
and discussed below. The mechanism of action of these drugs includes Hb
F induction, inhibition of cellular adhesion, anti-inflammatories,
surfactants, anti-platelets, vasodilators, anti-adhesives, inhibition
of Hb S polymerization, etc. It is rather unfortunate that the majority
of these drugs as well as HU were developed for indications other than
SCD. This is unlike other rare diseases such as hemophilia and cystic
fibrosis for which a few, if any, repurposed drugs are used. The
reasons for this disparity are not known. The complex pathophysiology
of SCD, its protean clinical manifestations and the suboptimal interest
from funders and scientists may be some of the reasons.
Potential Pharmacotherapeutic Drugs for the Treatment of SCDl
a. Targeting Hb F production:
Decitabine is an intravenous cytosine analog 5-aza-2’-deoxycytidine,
which hypomethylates DNA by inhibiting DNA methyltransferase. It is
approved for treatment of myelodysplastic syndrome. It increases fetal
Hb by reactivating the silenced γ-globin through hypomethylation at its
promoter site. In a small study of eight patients refractory or
intolerant to HU, it increased Hb F and Hb levels when administered
subcutaneously.[106] Ongoing trials will further
clarify its efficacy and tolerability. A Phase II study with planned
enrollment of 40 patients with high-risk SCD is recruiting.[107] A Phase I combination study of oral decitabine with tetrahydrouridine,[108]
a competitive inhibitor of cytidine deaminase, is also recruiting and
its aim is to evaluate oral bioavailability of decitabine in
combination therapy.[109,110]
Pomalidomide
is an orally active thalidomide analog developed by Celgene for the
treatment of graft versus host disease, SCA, myelofibrosis, scleroderma
and idiopathic pulmonary fibrosis. Preclinical studies showed that it
induced Hb F production in an SCD model with similar efficacy as HU.
Surprisingly, pomalidomide improved erythropoiesis in comparison to
myelosuppression seen with HU. However, when given in combination with
HU, this effect was lost and fetal Hb levels were suppressed.[111] A Phase I study of pomalidomide in SCD was completed. Twelve patients enrolled and data have not been published.[112]Panobinostat is a recently approved histone deacetylase (HDAC) inhibitor.[113] A study of panobinostat in patients with SCD is active but not recruiting yet.[114]
L-arginine, a substrate for NO, was evaluated in combination with HU in
a small randomized trial of 21 adult patients with SCD. There was a
greater response in fetal Hb levels and reticulocyte count in the group
receiving combination therapy versus HU alone. This study suggests that
fetal Hb synthesis depends on NO effect on erythroid progenitors.[115]b. Targeting adhesion: Intravenous Ig (IVIg) also inhibits leukocyte adhesion and activation by binding to FcγRIII expressed on neutrophils.[116]
A Phase I/II trial is currently recruiting to evaluate Gamunex
(Intravenous gamma globulin) versus normal saline in sickle cell acute
pain.[117]Low-molecular weight heparins (LMWH).
In a randomized clinical trial of 253 patients, Tinzaparin, an LMWH,
showed reduced duration of VOC and no severe bleeding complications.[118]
These results need to be validated in a multicenter study. A recent
Phase II trial of an oral P-selectin inhibitor (pentosan polysulfate
sodium) similar to heparin but with greater P-selectin blocking ability
than heparin showed improved microvascular flow in SCD patients in a
Phase I study.[119] Another LMWH, Dalteparin, was used in a completed phase II trial.[120]Crizanlizumab.
The efficacy of SelG1 (Crizanlizumab), a humanized anti-P-selectin
monoclonal antibody, in preventing VOC was evaluated in five different
trials. The first was the successful SUSTAIN trial that was approved by
the FDA on November 15, 2019 as described above. The remaining four
trials are as follows: •
The STAND trial whose purpose is to compare the efficacy and
safety of 2 doses of crizanlizumab (5.0 mg/kg and 7.5 mg/kg) versus
placebo in adolescent and adult SCD patients with a history of VOCs
leading to healthcare visit.[121]• The SPARTAN trial to evaluate the safety and efficacy of crizanlizumab in SCD related priapism.[122]•
Phase II CSEG101B2201 study is to confirm and to establish the
appropriate dosing and to evaluate the safety in pediatric patients
ages 6 months to <18 years with a history of VOC with or without HU,
receiving ranibizumab for 2 years. The approach is to extrapolate from
the PK/pharmacodynamics already established in the adult population.
The study is designed as a Phase II, multicenter, open-label study.[123]•
Phase II multicenter open label study to determine the
pharmacokinectics and pharmacodynamics study of SEG101 (criznalizumab)
in SCD patients with VOCs.[124]Propranolol significantly
reduced RBC adhesion in a dose-dependent manner. Adverse events were
not severe, did not vary with the dose administered and no elevation in
heart rate was noted. These results imply that β-blockers have a
potential role in inhibiting RBC adhesion.[125] A
Phase II study of propranolol in SCD has been completed and no data
have been reported at the time that this manuscript was written.[126] c. Targeting inflammationRegadenoson.
In SCA patients there is increase in the number of activated Invariant
Natural Killer T (iNKT) cells. Regadenoson is an A2A receptor agonist
that reduces the iNKT cells activation and thus decreases inflammation (Figure 3).
It was developed by CV Therapeutics, now Gilead Sciences, as an adjunct
in cardiac perfusion imaging. A Phase I study in 27 adults with SCD
showed a 48% decrease in activation of iNKT cells compared to baseline
after Regadenoson was administered with no toxicities identified.[127]
Randomized phase 2 trial of Regadenoson for treatment of acute VOCs in
SCD did not reduce iNKT cell activation to a prespecified level when
administered to patients with SCD. Since iNKT cell activation was not
reduced, the benefit of iNKT cell-based therapies in SCD cannot be
determined.[128] Further studies may be needed.
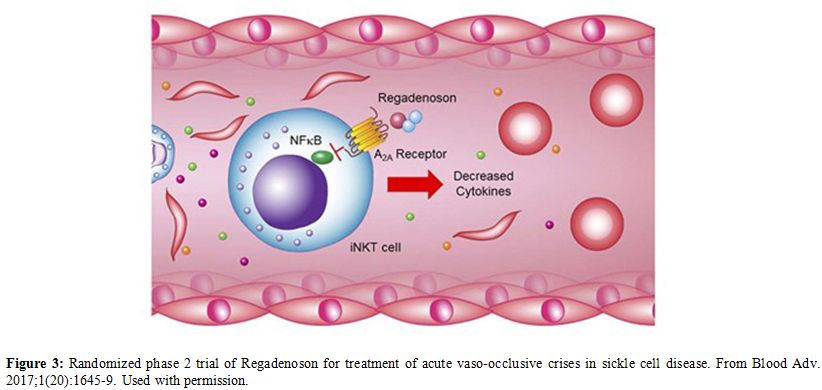 |
Figure 3. Randomized phase 2
trial of Regadenoson for treatment of acute vaso-occlusive crises in
sickle cell disease. From Blood Adv. 2017;1(20):1645-9. Used with
permission. |
NKTT-120
is an investigational drug developed by NKT Therapeutics to treat the
symptoms of SCA. It is a humanized monoclonal antibody designed to
target iNKT cells. Preclinical studies showed rapid and sustained iNKT
cell depletion in adults with SCD after the administration of NKTT-120.
Depletion of iNKT cells had no effect on other natural killer cells.
The T-cell antibody response was not impaired in response to a Keyhole
Limped Hemocyanin (KLH) challenge.[129] An
open-label, multi-center, single-ascending-dose study of NKTT120 to
determine its pharmacokinetics, pharmacodynamics and safety in patients
with SCA in the steady state showed rapid, specific and sustained iNKT
cell depletion without any toxicity or attributed serious adverse
events.[130]Statins.
The vascular injury seen in SCD has been described to share
similarities with that of atherosclerosis. Statins decrease
inflammation and improve endothelial function in cardiovascular disease
and are under study in SCD. They slow the production of cholesterol in
the body that may build up on the walls of the arteries and block blood
flow to the heart, brain, and other parts of the body. A pilot study of
26 patients treated with atorvastatin showed a dose-related decrease in
inflammatory biomarkers (C-reactive protein and IL-6 levels) and
increased NO metabolite levels.[131] A Phase II
trial of atorvastatin to determine its effect on blood vessels in
patients with SCD was first posted in November 2012. The primary
hypothesis is that endothelial dysfunction is an important contributor
to the pathophysiology of albuminuria in SCD. The investigators propose
that atorvastatin will improve endothelial dysfunction, decrease levels
of soluble fms-like tyrosine kinase-1 (sFLT-1), and decrease
albuminuria in patients with SCD. The study was completed on November
14, 2019. Results not available yet.[132]
Zileuton.
Sickle cell disease patients have elevated levels of 5-lipoxygenase, a
potent inflammatory leukotriene. Zileuton, a specific inhibitor of
5-lipoxygenase, is FDA approved for asthma. Beneficial effects in the
SCD animal model have led to a completed Phase I trial in SCD. It
showed that higher dose of Zileuton was safely tolerated by SCD
patients with good compliance.[133]N-acetylcysteine. N-acetylcysteine
(NAC) is an inexpensive amino acid derivative that replenishes
intracellular levels of the glutathione and it is the rate-limiting
substrate for glutathione generation, an important antioxidant with
pleiotropic effects on inflammation.[134] NAC
inhibits dense cell formation and restores glutathione levels toward
normal, which enables the cell to fight damage from ROS. It was used 30
years ago as a mucolytic agent in cystic fibrosis and asthma. In the
oral and parenteral routes, it treats acetaminophen toxicity. In pilot
studies, the administration of NAC resulted in a reduction of oxidative
stress. A Phase II, double-blind, randomized clinical trial was
completed to determine the efficacy of NAC in decreasing dense cell and
irreversible sickle cell formation and VOC episodes in SCD. NAC
inhibited dense cell formation, restored glutathione levels toward
normal and decreased VOC episodes.[135] A Phase III trial is underway.[136]Canakinumab.
Canakinumab has already been approved by the FDA in 2009 as ILARIS, an
interleukin-1β blocker indicated for the treatment of
Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children
4 years of age and older including: Familial Cold Autoinflammatory
Syndrome (FCAS) and Muckle-Wells Syndrome (MWS).[137]
Because of its anti-inflammatory potential it is being considered in a
study to determine its efficacy, safety and tolerability in pediatric
and young adult patients with SCA.[138]A
recent presentation at the 2019 American Society of Hematology annual
meeting described a multi-center, randomized, parallel group,
double-blind, placebo-controlled trial that recruited patients with SCA
(HbSS or HbS/ß0thalassemia) with history of ≥2 major pain
episodes/year, screening baseline detectable pain (using pain
e-diaries) and serum high sensitivity CRP level ≥1.0 mg/L. Patients
were randomized with 1:1 ratio to receive six monthly subcutaneous
injections of either canakinumab 300 mg (4 mg/kg for patients ≤40 kg)
or placebo. The concurrent use of hydroxyurea was a stratification
factor at randomization. Outcomes were measured at baseline and at
weeks 4, 8, 12, 16, 20, 24, after which all patients moved to open
label canakinumab treatment for additional 6 months.Interim
analysis for futility and safety was performed on the first 30 enrolled
patients (canakinumab, n=16; placebo, n=14), of whom 26 patients
completed the Week 12 assessments (canakinumab, n=14; placebo, n=12),
and 13 patients completed the Week 24 assessments. Enrolled patients
(median age 17 years, range 12-20; 19 males, 11 females) were evenly
distributed in the arms of the study. Results showed that Futility
criteria were not met and no canakinumab-associated safety issues were
identified in this first interim analysis. A second interim analysis is
pending.[139]Ambrisentan.
Ambriseentan (Letairis) is an endothelin receptor antagonist which has
already been approved by the FDA in 2007 for the treatment of pulmonary
arterial hypertension (PAH) (WHO Group 1): To improve exercise ability
and delay clinical worsening. In combination with tadalafil to
reduce the risks of disease progression and hospitalization for
worsening PAH, and to improve exercise ability. Preliminary data about
its potential role in SCD suggest that These data suggest that
endothelin receptor blockade is safe, well tolerated and has the
potential to impact various aspects of disease pathophysiology in SCD.[140-142]d. Targeting oxidative Injuryα-Lipoic acid.
Alpha-lipoic acid (ALA) is a potent antioxidant that is employed in the
treatment of several diseases. It augments cellular stress response by
increasing the transcription of antioxidant genes, decreasing NF-kB,
and increasing glutathione synthesis. Acetyl-l-carnitine is an
essential nutrient that facilitates the entry of long-chain fatty acids
into the mitochondria and decreases lipid peroxidation in tissue.
α-Lipoic acid and acetyl-L-carnitine have a synergistic antioxidant
effect.[143] A recent Phase II trial combining
antioxidants enrolled 42 patients to determine whether α-lipoic acid
and acetyl-L-carnitine will lower systemic inflammation in patients
with SCD. This study is complete; however, data is not available for
review.[144] In an open randomized trial ALA
treatment protected normal individuals from oxidative damage to lipids
and proteins. In SCD patients, the dose applied were not effective to
prevent the oxidative damage.[145] Further trials are not planned at the present.e. Targeting anti-coagulationRivaroxaban. The
direct oral anticoagulants (DOACs) include Rivaroxaban. Investigational
therapies targeting multiple pathways are being studied for the
treatment of SCD. Rivaroxaban, an orally active direct Factor-Xa
inhibitor and serine protease inhibitor, was FDA approved in the US as
an anticoagulant for prophylaxis and treatment in acute coronary
syndromes, cerebral ischemia, pulmonary embolism and venous thrombosis.
It is currently being evaluated in a Phase II clinical trial in SCD to
reduce inflammation, coagulation and endothelial cell activation, and
improve microvascular blood flow in patients during the non-VOC steady
state.[146]f. Targeting vasodilatation.Arginine.
Arginine is depleted in hemolysis due to the release of arginase and
leads to decreased NO formation. In SCD patients with pulmonary
hypertension, arginine supplementation increases plasma NO and rapidly
decreases pulmonary artery pressure by 15%.[147] A
recent randomized, double-blind, placebo-controlled study of high-dose
arginine supplementation in hospitalized SCD patients with VOC was
completed and found a > 56% reduction in opioid use in patients
receiving arginine compared with controls.[148] A
Phase II, randomized trial in 38 children showed a significant
reduction in opioid use and lower pain scores at discharge in those
treated with arginine in comparison to the placebo arm. There was no
significant difference in hospital length of stay and no toxicity was
noted.[149] A study was completed in children with
SCD to evaluate the effectiveness of arginine at increasing NO levels,
improving RBC function and reducing hospitalizations and pain
medication use. This was done by measuring gardos channel activity,
mean corpuscular Hb concentration (MCHC) and NO levels. There was only
statistically significant difference in low-dose arginine with
decreased MCHC versus placebo. Data is available but has not been
published.[150] Other studies have been completed and awaiting analysis and two are currently recruiting.[151-154]
Inhaled NO. As mentioned before NO failed as a therapeutic agent for hospitalized patients with SCD and VOC.[155]
Interestingly, the use of inhaled NO in the emergency department
significantly reduced pain scores compared with placebo (P < 0.02)
at the end of NO inhalation although both groups had similar baseline
pain scores.[156,157] Moreover, NO has been reported to reduces sickle Hb polymerization.[158]PF 04447943 (Phosphodiesterase 9A Inhibitor). A randomized, double-blinded, Phase 1b trial [159]
at 18 centers in the U.S. and Europe evaluated the safety and
tolerability of PF-04447943 over 29 days in people with stable SCD.
Multiple doses of PF-04447943, with or without HU, administered to
patients with SCD were generally well tolerated and showed
pharmacodynamics parameters suggestive of a protective effect against
vaso-occlusion. In addition, possible biomarkers to measure efficacy
for use in future SCD studies were noted.[160] Inhibition of PDE9A is required to treat diseases that lower the level of cGMP which, in turn, regulates signal transduction[161] and mediates vasodilatation.IMR-687
is a highly selective, potent inhibitor of phosphodiesterase 9. It has
a multimodal mechanism of action that acts primarily on RBC and has the
potential to act on white blood cells, adhesion mediators and other
cell types that are implicated in SCD. Currently, it is an
open-label extension study in adult patients with SCA who were
previously participants in the Phase 2a study titled "A Phase 2a,
Randomized, Double-Blind, Placebo-Controlled Study of IMR-687 in Adult
Patients with SCA".[162] This open-label extension
study will evaluate the long-term safety and tolerability of IMR-687 in
adult SCA patients. Exploratory long-term parameters will also be
examined. Riociguat
is used in a Phase 2 multi-center, randomized, double-blind,
placebo-controlled, parallel groups study aimed to evaluate its safety,
tolerability and efficacy compared with placebo in patients with SCD.[163]Olinciguat
is use in the STRONG SCD in patients with SCD. The primary aim of the
study is to evaluate the safety and tolerability of different dose
levels of Olinciguat compared with placebo when administered daily for
approximately 12 weeks to patients with stable SCD. Exploratory
objectives include evaluation of pharmacokinetic as well as evaluation
of its effect on symptoms of SCD, health-related quality of life, and
biomarkers of pharmacodynamic activity.[164]g. Targeting PolymerizationVoxelotor (OXBRYTA),
previously known as GBT440, has the potential to selectively bind to
Hb, and increase its affinity for oxygen. It also inhibits Hb
polymerization and prevents RBCS from becoming deformed. This should
restore normal RBC function and oxygen delivery. It should also help
reduce the risk of VOCs caused by sickle cells blocking blood vessels.Voxelotor
is oral, once-daily drug that binds to the α-chain of HbS, stabilizing
the molecule in the R-state conformation, which is known to interrupt
HbS polymerization.[101,165,166]
The target for HbS modification with voxelotor is 20%-30%. In phase 1/2
trials, Voxelotor inhibited HbS polymerization, RBC sickling, and
hemolysis, with a consequent increase in Hb concentration, while also
demonstrating an acceptable safety profile and was well tolerated.[167]
Phases 1/2 completed and Phase 3 randomized, placebo-controlled HOPE
trial involving patients with SCD, Voxelotor (1500 mg and 900 mg)
significantly increased and sustained Hb levels compared to placebo and
reduced markers of hemolysis. These findings are consistent with
inhibition of HbS polymerization and indicate a disease-modifying
potential. The secondary endpoints pertaining to frequency of VOC,
hospitalization stay, etc. we’re not significantly different from
placebo. Moreover, exploratory post-hoc trial showed that Voxelotor
resolved or improved leg ulcers in some patients. The new drug
application (NDA) for Voxelotor is currently under priority review by
the FDA which provides for a six-month review, and has been assigned a
Prescription Drug User Fee Act (PDUFA) target action date of February
26, 2020.Besides the HOPE trial, Voxelotor is being considered for other future trials. These include the following:•
Hemoglobin oxygen affinity modulation to inhibit Hb S
polymerization (HOPE-KIDS 2, GBT 440-032) trial. The objective of this
trial is to investigate the effect of Voxelotor on Transcranial Doppler
(TCD) flow velocity in pediatric patients with SCD with conditional TCD.• Actigraphy
improvement with Voxelotor (Active) trial. The objective of this trial
is to assess the impact of Voxelotor on physical activity, sleep
quality, and overall patient wellbeing in individuals with SCD. Part 1
of this trial will be a phase 4 open-label, single-arm, within-subject
comparison followed by Part 2 trial which is a randomized withdrawal
placebo-controlled trial. FT-4202 (PKR Activator).
FT-4202 is a selective RBC pyruvate kinase-R activator (PKR) to be used
as a modifying therapy for the treatment of SCD. Its mechanism of
action includes activating the RBC’s natural PKR activity to decrease
2,3-DPG levels which results in shifting the oxygen dissociation curve
to the left causing Hb to hold on to oxygen molecules longer to
decrease RBC sickling. In addition, the downstream action of FT-4202
increases ATP levels that provide energy to RBCs health and survival.
These effects would increase Hb levels and possibly decrease the
frequency of VOCs.[168,169]h. Targeting SupplementsNiacin (Vitamin B3).
Niacin is a drug that has been used to increase high density
cholesterol (HDL), the “good cholesterol”. It improves the blood flow
in people with SCD.[170]Niacin,
a drug that has been used to increase HDL (good cholesterol) levels,
improves blood flow in people without SCD. This study will see if it
can do the same in people with the disease.Cholecalciferol (Vitamin D3). About
98% of patients with SCD have vitamin D deficiency, defined as a
25-hydroxyvitamin D level (25(OH)D) less than or equal to 20 ng/mL. As
a result of low bone density, patients may develop osteonecrosis,
chronic inflammation and related pain.[171] Since
vitamin D regulates calcium levels and supports bone health, its
deficiency may worsen musculoskeletal health problems already present
in people with SCD. However, a Cochrane review study showed that the
evidence for vitamin D3 supplementation in patients with SCD is not of
sufficient quality to guide clinical practice. Evidence of vitamin D
supplementation in sickle cell disease from high quality studies is
needed.[172].
Conclusions
There
has been tremendous advance in our knowledge of the pathophysiology of
sickle cell vascular injury over the past decade resulting in new
therapeutic targets. The field is witnessing promising translational
studies hoping to replace or use with HU as the primary pharmacologic
therapy for patients with SCD. This review includes therapies targeting
increases in fetal Hb and the complex pathways in adhesion,
inflammation, oxidative damage and polymerization.
Hydroxyurea is
an oral agent that has decreased morbidity and mortality in adults and
children with SCA. It decreases recurrent VOCs, ACS and blood
transfusion requirements, and improves quality of life mainly through
increasing fetal Hb production. It is inexpensive and potentially
available worldwide. It is cytotoxic, which may cause myelosuppression
and its carcinogenic effects are unknown and long-term studies
have failed to document this. Traditionally, it has been.
contraindicated in pregnancy and during lactation due to potential
teratogenicity. Recent anecdotes and case reports indicated its safety
during pregnancy and lactation. Its role in pregnancy and lactation is
currently the subject of clinical trials. It seems it should not be
taken during the first two trimesters of pregnancy.
L-glutamine is
metabolized to glutamate, the glutathione precursor, and preserves
intracellular NAD, which is necessary for glutathione recycling. Oral
supplementation of glutamine in SCD increases the NAD redox potential
and may improve sickle erythrocyte adhesiveness. Oral glutamine was
developed by Emmaus Medical for the treatment of short bowel syndrome
and in SCA and β thalassemia. It decreases the resting energy
expenditure in children with SCD. A multicenter Phase III trial of L-
glutamine supplementation in 230 children to prevent VOC is completed
Results showed that the median number of pain crises over 48 weeks was
lower among those who received oral therapy with L-glutamine,
administered alone or with HU, than among those who received placebo,
with or without HU.
Decitabine is an attractive agent as it
induced fetal Hb with similar disadvantageous risk profile like HU with
potential myelosuppression, teratogenicity and carcinogenicity. It is
an already approved therapy for myelodysplastic syndrome and acute
myeloid leukemia, conditions more prevalent in the elderly. It is being
evaluated in oral form and in combination therapy currently and further
testing is warranted in the pediatric population. Unlike HU, its effect
to increase Hb F level occurs much sooner than that for HU.
N-acetylcysteine has reached Phase III trials. It targets inflammation.
A combination with a fetal Hb-inducing agent such as HU is a potential
strategy to combat SCD. Studies involving NO so far have been
disappointing in the sickle cell population. It is surprising that
arginine therapy. was more promising than NO since its role is to
increase NO. Nevertheless, this natural amino acid is an ideal agent
for a combination regimen.
In the sickle cell population, there
are challenges with clinical trial enrollment since it is a relatively
rare and clinically heterogeneous disease. A paradigm shift in clinical
trial design would improve outcome. Due to the complex pathophysiology
of the disease, clinical trials targeting a multi-agent approach may be
more successful as in oncology where combination chemotherapy regimens
have been more efficacious. Trial design in SCD over the past three
decades has historically incorporated all patients with SCA. Recently,
this approach is being modified to reassess endpoints to determine
benefits in targeted phenotypes, including quality-of-life measures and
incorporating biomarkers in patient selection.
In summary, our
greater understanding of the pathophysiology of SCD has led to many new
targets for drug therapy, and with a paradigm shift in clinical trial
design. We are in an exciting position to improve care for the millions
who suffer from SCD. It is very probable that in the near future we may
witness new trials to treat SCD that contain two or more drugs that
have different mechanism of action. My prediction is that such trials
may have acronyms such as FOC, FOV, FOCV, etc. trials where F refers to
a drug that increases Hb F, O refers to an antioxidant drug, C refers
to anti-adhesion drug and V to anti-polymerization drug or other
possible combinations.
References
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams
TN. Global burden of sickle cell anaemia in children under five,
2010-2050: modelling based on demographics, excess mortality, and
interventions. PLoS Med. 2013;10(7):e1001484. https://doi.org/10.1371/journal.pmed.1001484 PMid:23874164 PMCid:PMC3712914
- Odame
I. Developing a global agenda for sickle cell disease: report of an
international symposium and workshop in Cotonou, Republic of Benin. Am
J Prev Med. 2010;38(4 Suppl):S571-5. https://doi.org/10.1016/j.amepre.2009.12.021 PMid:20331960
- McGann
PT, Ware RE. Hydroxyurea for sickle cell anemia: what have we learned
and what questions still remain? Curr Opin Hematol. 2011;18(3):158-65. https://doi.org/10.1097/MOH.0b013e32834521dd PMid:21372708 PMCid:PMC3181131
- Castro
O, Rana SR, Bang KM, Scott RB. Age and prevalence of sickle-cell trait
in a large ambulatory population. Genet Epidemiol. 1987;4(4):307-11. https://doi.org/10.1002/gepi.1370040409 PMid:3666437
- Steinberg
M, H., Forget BG, Higgs D, R., Weatherall DJ. Disorders of hemoglobin:
Genetics, Pathophysiology, and Clinical Management, Second Edition. 2nd ed. Cambridge: Cambridge University Press; 2009. 826 p. https://doi.org/10.1017/CBO9780511596582
- Goldsmith
JC, Bonham VL, Joiner CH, Kato GJ, Noonan AS, Steinberg MH. Framing the
research agenda for sickle cell trait: building on the current
understanding of clinical events and their potential implications. Am J
Hematol. 2012;87(3):340-6. https://doi.org/10.1002/ajh.22271 PMid:22307997 PMCid:PMC3513289
- Serjeant GR, Serjeant BE. Sickle cell disease, 3rd edition. Oxford: Oxfird University Press; 2001. 772 p. https://doi.org/10.1046/j.1365-2141.2001.02557.x PMid:11167776
- Bunn HF, Forget BG. Hemoglobin: Molecular, Genetic and Clinical Aspects. Philadelphia: WB Saunders; 1986.
- Ballas
SK, Park D, Wapner RJ. Neonatal screening for sickle cell disease in a
metropolitan university hospital: efficacy and problems. J Med Screen.
1994;1(4):229-32. https://doi.org/10.1177/096914139400100409 PMid:8790526
- Shafer
FE, Lorey F, Cunningham GC, Klumpp C, Vichinsky E, Lubin B. Newborn
screening for sickle cell disease: 4 years of experience from
California's newborn screening program. J Pediatr Hematol Oncol.
1996;18(1):36-41. https://doi.org/10.1097/00043426-199602000-00007 PMid:8556368
- Diallo
DA. Sickle cell disease in Africa: current situation and strategies for
improving the quality and duration of survival. Bull Acad Natl Med.
2008;192(7):1361-72; discussion 72-3.
- Vichinsky
E. Emerging 'A' therapies in hemoglobinopathies: agonists, antagonists,
antioxidants, and arginine. Hematology Am Soc Hematol Educ Program.
2012;2012:271-5. https://doi.org/10.1182/asheducation.V2012.1.271.3798318 PMid:23233591
- Ballas SK. Sickle Cell Pain, 2nd Edition. Washington DC: International Association for the Study of Pain; 2014.
- Zhang
D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and
inflammatory pathways at the nexus of sickle cell disease
pathophysiology. Blood. 2016;127(7):801-9. https://doi.org/10.1182/blood-2015-09-618538 PMid:26758915 PMCid:PMC4760086
- Motta
I, Ghiaccio V, Cosentino A, Breda L. Curing Hemoglobinopathies:
Challenges and Advances of Conventional and New Gene Therapy
Approaches. Mediterr J Hematol Infect Dis. 2019 Nov 1;11(1):e2019067.
doi: 10.4084/MJHID.2019.067. eCollection 2019. Review. PubMed PMID:
31700592; PubMed Central PMCID:PMC6827604.
- Ballas SK. Sickle cell anaemia: progress in pathogenesis and treatment. Drugs. 2002;62(8):1143-72. https://doi.org/10.2165/00003495-200262080-00003 PMid:12010077
- Kotiah SD, Ballas SK. Investigational drugs in sickle cell anemia. Expert Opin Investig Drugs. 2009;18(12):1817-28. https://doi.org/10.1517/13543780903247463 PMid:19780709
- Kauf
TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health
care for children and adults with sickle cell disease. Am J Hematol.
2009;84(6):323-7. https://doi.org/10.1002/ajh.21408 PMid:19358302
- Lanzkron
S, Haywood C, Segal JB, Dover GJ. Hospitalization rates and costs of
care of patients with sickle-cell anemia in the state of Maryland in
the era of hydroxyurea. Am J Hematol. 2006;81(12):927-32. https://doi.org/10.1002/ajh.20703 PMid:16924648
- Moore
RD, Charache S, Terrin ML, Barton FB, Ballas SK. Cost-effectiveness of
hydroxyurea in sickle cell anemia. Investigators of the Multicenter
Study of Hydroxyurea in Sickle Cell Anemia. Am J Hematol.
2000;64(1):26-31. https://doi.org/10.1002/(SICI)1096-8652(200005)64:1<26::AID-AJH5>3.0.CO;2-F
- Benjamin
LJ, Swinson GI, Nagel RL. Sickle cell anemia day hospital: an approach
for the management of uncomplicated painful crises. Blood.
2000;95(4):1130-6. https://doi.org/10.1182/blood.V95.4.1130.003k03a_1130_1136 PMid:10666181
- Ballas SK. The cost of health care for patients with sickle cell disease. Am J Hematol. 2009;84(6):320-2. https://doi.org/10.1002/ajh.21443 PMid:19415728
- Manwani
D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology
and novel targeted therapies. Hematology Am Soc Hematol Educ Program.
2013;2013:362-9. https://doi.org/10.1182/asheducation-2013.1.362 PMid:24319205
- Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. 2009;16(1):97-111. https://doi.org/10.1080/10739680802279394 PMid:18720225 PMCid:PMC3059190
- Madigan
C, Malik P. Pathophysiology and therapy for haemoglobinopathies. Part
I: sickle cell disease. Expert Rev Mol Med. 2006;8(9):1-23. https://doi.org/10.1017/S1462399406010659
- Gutsaeva
DR, Parkerson JB, Yerigenahally SD, Kurz JC, Schaub RG, Ikuta T, et al.
Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential
therapeutic agent for sickle cell disease. Blood. 2011;117(2):727-35. https://doi.org/10.1182/blood-2010-05-285718 PMid:20926770 PMCid:PMC3031491
- Turhan
A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for
adherent leukocytes in sickle cell vascular occlusion: a new paradigm.
Proc Natl Acad Sci U S A. 2002;99(5):3047-51. https://doi.org/10.1073/pnas.052522799 PMid:11880644 PMCid:PMC122470
- Zennadi
R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, Shan S, et al.
Epinephrine-induced activation of LW-mediated sickle cell adhesion and
vaso-occlusion in vivo. Blood. 2007;110(7):2708-17. https://doi.org/10.1182/blood-2006-11-056101 PMid:17609430 PMCid:PMC1988948
- Hines
PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, et al. Novel
epinephrine and cyclic AMP-mediated activation of BCAM/Lu-dependent
sickle (SS) RBC adhesion. Blood. 2003;101(8):3281-7. https://doi.org/10.1182/blood-2001-12-0289 PMid:12506027
- Schaer
DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and
free hemoglobin revisited: exploring hemoglobin and hemin scavengers as
a novel class of therapeutic proteins. Blood. 2013;121(8):1276-84. https://doi.org/10.1182/blood-2012-11-451229 PMid:23264591 PMCid:PMC3578950
- Morris
CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al.
Dysregulated arginine metabolism, hemolysis-associated pulmonary
hypertension, and mortality in sickle cell disease. JAMA.
2005;294(1):81-90. https://doi.org/10.1001/jama.294.1.81 PMid:15998894 PMCid:PMC2065861
- Kato
GJ, Wang Z, Machado RF, Blackwelder WC, Taylor JGt, Hazen SL.
Endogenous nitric oxide synthase inhibitors in sickle cell disease:
abnormal levels and correlations with pulmonary hypertension,
desaturation, haemolysis, organ dysfunction and death. Br J Haematol.
2009;145(4):506-13. https://doi.org/10.1111/j.1365-2141.2009.07658.x PMid:19344390 PMCid:PMC2935697
- Chantrathammachart P, Pawlinski R. Tissue factor and thrombin in sickle cell anemia. Thromb Res. 2012;129 Suppl 2:S70-2. https://doi.org/10.1016/j.thromres.2012.02.038 PMid:22398014 PMCid:PMC3335974
- Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(1):72-80. https://doi.org/10.1002/iub.584 PMid:22131167
- Gizi
A, Papassotiriou I, Apostolakou F, Lazaropoulou C, Papastamataki M,
Kanavaki I, et al. Assessment of oxidative stress in patients with
sickle cell disease: The glutathione system and the oxidant-antioxidant
status. Blood Cells Mol Dis. 2011;46(3):220-5. https://doi.org/10.1016/j.bcmd.2011.01.002 PMid:21334230
- Nur
E, Biemond BJ, Otten HM, Brandjes DP, Schnog JJ. Oxidative stress in
sickle cell disease; pathophysiology and potential implications for
disease management. Am J Hematol. 2011;86(6):484-9. https://doi.org/10.1002/ajh.22012 PMid:21544855
- Steinberg
MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The
risks and benefits of long-term use of hydroxyurea in sickle cell
anemia: A 17.5 year follow-up. Am J Hematol. 2010;85(6):403-8. https://doi.org/10.1002/ajh.21699 PMid:20513116 PMCid:PMC2879711
- Expert
Panel Report. Evidence-Based Management of Sickle Cell Disease Bethesda
MD: National Heart, Lung, and Blood Institute; 2014 [Available from: http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines/].
- Ballas
SK, McCarthy WF, Guo N, DeCastro L, Belevue R, Barton BA, et al.
Exposure to hydroxyurea and pregnancy outcomes in patients with sickle
cell anemia. J Natl Med Assoc. 2009;101(10):1046-51. https://doi.org/10.1016/S0027-9684(15)31072-5
- Borba
R, Lima CS, Grotto HZ. Reticulocyte parameters and hemoglobin F
production in sickle cell disease patients undergoing hydroxyurea
therapy. J Clin Lab Anal. 2003;17(2):66-72. https://doi.org/10.1002/jcla.10070 PMid:12640630 PMCid:PMC6807693
- Guarda
CC, Silveira-Mattos PSM, Yahouedehou S, Santiago RP, Aleluia MM,
Figueiredo CVB, et al. Hydroxyurea alters circulating monocyte subsets
and dampens its inflammatory potential in sickle cell anemia patients.
Sci Rep. 2019;9(1):14829. https://doi.org/10.1038/s41598-019-51339-x PMid:31616024 PMCid:PMC6794261
- Penkert
RR, Hurwitz JL, Thomas P, Rosch J, Dowdy J, Sun Y, et al. Inflammatory
molecule reduction with hydroxyurea therapy in children with sickle
cell anemia. Haematologica. 2018;103(2):e50-e4. https://doi.org/10.3324/haematol.2017.177360 PMid:29146708 PMCid:PMC5792285
- Ballas
SK, Connes P. Rheological properties of sickle erythrocytes in patients
with sickle-cell anemia: The effect of hydroxyurea, fetal hemoglobin,
and alpha-thalassemia. Eur J Haematol. 2018;101(6):798-803. https://doi.org/10.1111/ejh.13173 PMid:30204261 PMCid:PMC6224298
- Torres
Lde S, da Silva DG, Belini Junior E, de Almeida EA, Lobo CL, Cancado
RD, et al. The influence of hydroxyurea on oxidative stress in sickle
cell anemia. Rev Bras Hematol Hemoter. 2012;34(6):421-5. https://doi.org/10.5581/1516-8484.20120106 PMid:23323065 PMCid:PMC3545428
- Gardner
K, Bell C, Bartram JL, Allman M, Awogbade M, Rees DC, et al. Outcome of
adults with sickle cell disease admitted to critical care - experience
of a single institution in the UK. Br J Haematol. 2010;150(5):610-3. https://doi.org/10.1111/j.1365-2141.2010.08271.x PMid:20560967
- Brewin
J, Tewari S, Menzel S, Kirkham F, Inusa B, Renney G, et al. The effects
of hydroxycarbamide on the plasma proteome of children with sickle cell
anaemia. Br J Haematol. 2019;186(6):879-86. https://doi.org/10.1111/bjh.15996 PMid:31140594
- Dasgupta
T, Fabry ME, Kaul DK. Antisickling property of fetal hemoglobin
enhances nitric oxide bioavailability and ameliorates organ oxidative
stress in transgenic-knockout sickle mice. Am J Physiol Regul Integr
Comp Physiol. 2010;298(2):R394-402. https://doi.org/10.1152/ajpregu.00611.2009 PMid:20007516 PMCid:PMC2828175
- Kaul
DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on
microvascular regulation in sickle transgenic-knockout mice. J Clin
Invest. 2004;114(8):1136-45. https://doi.org/10.1172/JCI200421633 PMid:15489961 PMCid:PMC522244
- Rees DC. The rationale for using hydroxycarbamide in the treatment of sickle cell disease. Haematologica. 2011;96(4):488-91. https://doi.org/10.3324/haematol.2011.041988 PMid:21454878 PMCid:PMC3069221
- Davies S, Olujohungbe A. Hydroxyurea for sickle cell disease. Cochrane Database Syst Rev. 2001(2):CD002202.
- Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358(13):1362-9. https://doi.org/10.1056/NEJMct0708272 PMid:18367739
- Ballas
SK, Marcolina MJ, Dover GJ, Barton FB. Erythropoietic activity in
patients with sickle cell anaemia before and after treatment with
hydroxyurea. Br J Haematol. 1999;105(2):491-6. https://doi.org/10.1111/j.1365-2141.1999.01339.x PMid:10233426
- Charache
S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect
of hydroxyurea on the frequency of painful crises in sickle cell
anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle
Cell Anemia. N Engl J Med. 1995;332(20):1317-22. https://doi.org/10.1056/NEJM199505183322001 PMid:7715639
- Charache
S, Barton FB, Moore RD, Terrin ML, Steinberg MH, Dover GJ, et al.
Hydroxyurea and sickle cell anemia. Clinical utility of a
myelosuppressive "switching" agent. The Multicenter Study of
Hydroxyurea in Sickle Cell Anemia. Medicine. 1996;75(6):300-26. https://doi.org/10.1097/00005792-199611000-00002 PMid:8982148
- Alvarez
O, Miller ST, Wang WC, Luo Z, McCarville MB, Schwartz GJ, et al. Effect
of hydroxyurea treatment on renal function parameters: results from the
multi-center placebo-controlled BABY HUG clinical trial for infants
with sickle cell anemia. Pediatr Blood Cancer. 2012;59(4):668-74. https://doi.org/10.1002/pbc.24100 PMid:22294512 PMCid:PMC3396762
- Armstrong
FD, Elkin TD, Brown RC, GlassP, Rana S, Casella JF, et al.
Developmental function in toddlers with sickle cell anemia. Pediatrics.
2013;131(2):e406-14. https://doi.org/10.1542/peds.2012-0283 PMid:23296434 PMCid:PMC3557401
- Lebensburger
JD, Miller ST, Howard TH, Casella JF, Brown RC, Lu M, et al. Influence
of severity of anemia on clinical findings in infants with sickle cell
anemia: analyses from the BABY HUG study. Pediatr Blood Cancer.
2012;59(4):675-8. https://doi.org/10.1002/pbc.24037 PMid:22190441 PMCid:PMC3337342
- McGann
PT, Flanagan JM, Howard TA, Dertinger SD, He J, Kulharya AS, et al.
Genotoxicity associated with hydroxyurea exposure in infants with
sickle cell anemia: results from the BABY-HUG phase III clinical trial.
Pediatr Blood Cancer. 2012;59(2):254-7. https://doi.org/10.1002/pbc.23365 PMid:22012708 PMCid:PMC3277805
- Wang
WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al.
Hydroxycarbamide in very young children with sickle-cell anaemia: a
multicentre, randomised, controlled trial (BABY HUG). Lancet.
2011;377(9778):1663-72. https://doi.org/10.1016/S0140-6736(11)60355-3
- Lam
MS. Extemporaneous compounding of oral liquid dosage formulations and
alternative drug delivery methods for anticancer drugs.
Pharmacotherapy. 2011;31(2):164-92. https://doi.org/10.1592/phco.31.2.164 PMid:21275495
- Ballas
SK, Singh P, Adams-Graves P, Wordell CJ. Idiosyncratic Side Effects of
Hydroxyurea in Patients with Sickle Cell Anemia. J Blood Disorders
Transf. 2013;4:5.
- Su ZT, Segal JB,
Lanzkron S, Ogunsile FJ. National trends in hydroxyurea and opioid
prescribing for sickle cell disease by office-based physicians in the
United States, 1997-2017. Pharmacoepidemiol Drug Saf.
2019;28(9):1246-50. https://doi.org/10.1002/pds.4860 PMid:31328369
- Schuchard
SB, Lissick JR, Nickel A, Watson D, Moquist KL, Blaylark RM, et al.
Hydroxyurea use in young infants with sickle cell disease. Pediatr
Blood Cancer. 2019;66(7):e27650. https://doi.org/10.1002/pbc.27650 PMid:30729675
- Thomas
R, Dulman R, Lewis A, Notarangelo B, Yang E. Prospective longitudinal
follow-up of children with sickle cell disease treated with hydroxyurea
since infancy. Pediatr Blood Cancer. 2019;66(9):e27816. https://doi.org/10.1002/pbc.27816 PMid:31157521
- Ware
RE, McGann PT, Quinn CT. Hydroxyurea for children with sickle cell
anemia: Prescribe it early and often. Pediatr Blood Cancer.
2019;66(8):e27778. https://doi.org/10.1002/pbc.27778 PMid:31038282
- Creary
SE, Modi AC, Stanek JR, Chisolm DJ, O'Brien SH, Nwankwo C, et al.
Allocation of Treatment Responsibility and Adherence to Hydroxyurea
Among Adolescents With Sickle Cell Disease. J Pediatr Psychol. 2019. https://doi.org/10.1093/jpepsy/jsz061 PMid:31403687
- Jabour
SM, Beachy S, Coburn S, Lanzkron S, Eakin MN. The Role of
Patient-Physician Communication on the Use of Hydroxyurea in Adult
Patients with Sickle Cell Disease. J Racial Ethn Health Disparities.
2019;6(6):1233-43. https://doi.org/10.1007/s40615-019-00625-5 PMid:31410784
- Mvalo
T, Topazian HM, Kamthunzi P, Chen JS, Kambalame I, Mafunga P, et al.
Real-world experience using hydroxyurea in children with sickle cell
disease in Lilongwe, Malawi. Pediatr Blood Cancer. 2019;66(11):e27954. https://doi.org/10.1002/pbc.27954 PMid:31397075
- Adeyemo
TA, Diaku-Akinwunmi IN, Ojewunmi OO, Bolarinwa AB, Adekile AD. Barriers
to the use of hydroxyurea in the management of sickle cell disease in
Nigeria. Hemoglobin. 2019;43(3):188-92. https://doi.org/10.1080/03630269.2019.1649278 PMid:31462098
- Ohene-Frempong
K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al.
Cerebrovascular accidents in sickle cell disease: rates and risk
factors. Blood. 1998;91(1):288-94.
- Hogan
AM, Vargha-Khadem F, Saunders DE, Kirkham FJ, Baldeweg T. Impact of
frontal white matter lesions on performance monitoring: ERP evidence
for cortical disconnection. Brain. 2006;129(Pt 8):2177-88. https://doi.org/10.1093/brain/awl160 PMid:16815874
- Pegelow
CH, Macklin EA, Moser FG, Wang WC, Bello JA, Miller ST, et al.
Longitudinal changes in brain magnetic resonance imaging findings in
children with sickle cell disease. Blood. 2002;99(8):3014-8. https://doi.org/10.1182/blood.V99.8.3014 PMid:11929794
- el
Gammal T, Adams RJ, Nichols FT, McKie V, Milner P, McKie K, et al. MR
and CT investigation of cerebrovascular disease in sickle cell
patients. AJNR Am J Neuroradiol. 1986;7(6):1043-9.
- Abboud
MR, Yim E, Musallam KM, Adams RJ. Discontinuing prophylactic
transfusions increases the risk of silent brain infarction in children
with sickle cell disease: data from STOP II. Blood. 2011;118(4):894-8. https://doi.org/10.1182/blood-2010-12-326298 PMid:21633086 PMCid:PMC3148169
- Scantlebury
N, Mabbott D, Janzen L, Rockel C, Widjaja E, Jones G, et al. White
matter integrity and core cognitive function in children diagnosed with
sickle cell disease. J Pediatr Hematol Oncol. 2011;33(3):163-71. https://doi.org/10.1097/MPH.0b013e3182036f33 PMid:21325970
- Wang
WC, Pavlakis SG, Helton KJ, McKinstry RC, Casella JF, Adams RJ, et al.
MRI abnormalities of the brain in one-year-old children with sickle
cell anemia. Pediatr Blood Cancer. 2008;51(5):643-6. https://doi.org/10.1002/pbc.21612 PMid:18478575
- Ware RE, Helms RW. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood. 2012;119(17):3925-32. https://doi.org/10.1182/blood-2011-11-392340 PMid:22318199 PMCid:PMC3350359
- Ware
RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al.
Hydroxycarbamide versus chronic transfusion for maintenance of
transcranial doppler flow velocities in children with sickle cell
anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a
multicentre, open-label, phase 3, non-inferiority trial. Lancet.
2016;387(10019):661-70. https://doi.org/10.1016/S0140-6736(15)01041-7
- Kapustin
D, Leung J, Odame I, Williams S, Shroff M, Kassner A. Hydroxycarbamide
treatment in children with Sickle Cell Anaemia is associated with more
intact white matter integrity: a quantitative MRI study. Br J Haematol.
2019;187(2):238-45. https://doi.org/10.1111/bjh.16063 PMid:31215028
- Hankins
JS, McCarville MB, Rankine-Mullings A, Reid ME, Lobo CL, Moura PG, et
al. Prevention of conversion to abnormal transcranial Doppler with
hydroxyurea in sickle cell anemia: A Phase III international randomized
clinical trial. Am J Hematol. 2015;90(12):1099-105. https://doi.org/10.1002/ajh.24198 PMid:26414435 PMCid:PMC4715740
- Sirieix
ME, Debure C, Baudot N, Dubertret L, Roux ME, Morel P, et al. Leg
ulcers and hydroxyurea: forty-one cases. Arch Dermatol.
1999;135(7):818-20. https://doi.org/10.1001/archderm.135.7.818 PMid:10411157
- Koshy M, Enstuah R, Koranda A. Leg ulcers in patients in sickle cell disease. Blood. 1989;74:1403-8. https://doi.org/10.1182/blood.V74.4.1403.1403 PMid:2475188
- Soya
E, Makowski C, Blaise S. Leg ulcer induced by hydroxycarbamide in
sickle cell disease: What is the therapeutic impact? Int Wound J.
2019;16(4):897-902. https://doi.org/10.1111/iwj.13115 PMid:30916480
- de
Montalembert M, Begue P, Bernaudin F, Thuret I, Bachir D, Micheau M.
Preliminary report of a toxicity study of hydroxyurea in sickle cell
disease. French Study Group on Sickle Cell Disease. Arch Dis Child.
1999;81(5):437-9. https://doi.org/10.1136/adc.81.5.437 PMid:10519721 PMCid:PMC1718114
- Byrd
DC, Pitts SR, Alexander CK. Hydroxyurea in two pregnant women with
sickle cell anemia. Pharmacotherapy. 1999;19(12):1459-62. https://doi.org/10.1592/phco.19.18.1459.30901 PMid:10600098
- Diav-Citrin
O, Hunnisett L, Sher GD, Koren G. Hydroxyurea use during pregnancy: a
case report in sickle cell disease and review of the literature. Am J
Hematol. 1999;60(2):148-50. https://doi.org/10.1002/(SICI)1096-8652(199902)60:2<148::AID-AJH12>3.0.CO;2-I
- Children's
Hospital Medical Center Cincinnati. Hydroxyurea Exposure Limiting
Pregnancy and Follow-Up Lactation (HELPFUL) (NCT04093986):
ClinicalTrials.gov; [Available from: https://clinicaltrials.gov/ct2/show/NCT04093986 (Accessed on November 18, 2019)].
- Pistilli
B, Bellettini G, Giovannetti E, Codacci-Pisanelli G, Azim HA, Jr.,
Benedetti G, et al. Chemotherapy, targeted agents, antiemetics and
growth-factors in human milk: how should we counsel cancer patients
about breastfeeding? Cancer Treat Rev. 2013;39(3):207-11. https://doi.org/10.1016/j.ctrv.2012.10.002 PMid:23199900
- Ware
RE, Marahatta A, Ware JL, al. e. Hydroxyurea exposure in lactation-a
pharmacokinetics study (HELPS). Blood. 2018;132(Suppl 1):3677. https://doi.org/10.1182/blood-2018-99-114142
- Children's
Hospital Medical Center Cincinnati. Hydoxyurea Exposure in Lactation A
Pharmacokinetics Study (HELPS) (HELPS) (NCT02990598):
ClinicalTrials.gov; [Available from: https://clinicaltrials.gov/ct2/show/NCT02990598 (Accessed on November 18, 2019)].
- Brosnan JT. Interorgan amino acid transport and its regulation. J Nutr. 2003;133(6 Suppl 1):2068s-72s. https://doi.org/10.1093/jn/133.6.2068S PMid:12771367
- Niihara
Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A Phase 3
Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med.
2018;379(3):226-35. https://doi.org/10.1056/NEJMoa1715971 PMid:30021096
- Niihara Y, Smith WR, Stark CW. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. 2018;379(19):1880. https://doi.org/10.1056/NEJMoa1715971
- Minniti CP. l-Glutamine and the Dawn of Combination Therapy for Sickle Cell Disease. N Engl J Med. 2018;379(3):292-4. https://doi.org/10.1056/NEJMe1800976 PMid:30021091
- Emmaus
Medical Inc. A Phase III Safety and Efficacy Study of L-Glutamine to
Treat Sickle Cell Disease or Sickle βo-thalassemia (NCT01179217):
ClinicalTrials.gov; [Available from: https://clinicaltrials.gov/ct2/show/NCT01179217 (Accessed on November 18, 2019)].
- Emmaus
Medical Inc. L-Glutamine Therapy for Sickle Cell Anemia and Sickle ß0
Thalassemia (NCT00125788) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00125788 (Accessed on November 18, 2019)].
- St.
Jude Children's Research Hospital. Trial of Oral Glutamine in Patients
with Sickle Cell Anemia (NCT00131508): ClinicalTrials.gov; [Available
from: https://clinicaltrials.gov/ct2/show/NCT00131508 (Accessed on November 18, 2019)].
- Emmaus Medical Inc. ENDARI (L-glutamine oral powder) [Package insert]. Torrance, CA.2017.
- Ataga
KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al.
Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease.
N Engl J Med. 2017;376(5):429-39. https://doi.org/10.1056/NEJMoa1611770 PMid:27959701 PMCid:PMC5481200
- Novartis Pharmaceuticals. ADAKVEO (crizanlizumab-tmca) injection [Package insert]. East Hanover, NJ.2019.
- Vichinsky
E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A Phase
3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med.
2019;381(6):509-19. https://doi.org/10.1056/NEJMoa1903212 PMid:31199090
- Global
Blood Therapeutics. Study to Evaluate the Effect of Voxelotor
Administered Orally to Patients With Sickle Cell Disease (GBT_HOPE)
(GBT_HOPE) [NCT03036813] ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03036813 (Accessed on December 16, 2019)].
- Global Blood Therapeutics. OXBRYTA (voxelotor) tablets [Package insert]. San Francisco, CA.2019.
- GlycoMimetics,
editor GlycoMimetics Announces Presentation of Rivipansel Data in
Pediatric Patients at American Society of Pediatric Hematology Oncology
27th Annual Meeting 2014 May 15; Palmer House Hilton Hotel, Chicago.
- Telen
MJ, Wun T, McCavit TL, De Castro LM, Krishnamurti L, Lanzkron S, et al.
Randomized phase 2 study of GMI-1070 in SCD: reduction in time to
resolution of vaso-occlusive events and decreased opioid use. Blood.
2015;125(17):2656-64. https://doi.org/10.1182/blood-2014-06-583351 PMid:25733584 PMCid:PMC4408290
- Saunthararajah
Y, Hillery CA, Lavelle D, Molokie R, Dorn L, Bressler L, et al. Effects
of 5-aza-2'-deoxycytidine on fetal hemoglobin levels, red cell
adhesion, and hematopoietic differentiation in patients with sickle
cell disease. Blood. 2003;102(12):3865-70. https://doi.org/10.1182/blood-2003-05-1738 PMid:12907443
- National
Heart Lung and Blood Institute (NHLBI). Decitabine for High-Risk Sickle
Cell Disease (NCT01375608) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01375608 (Accessed on November 18, 2019)].
- Molokie
R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, et al. Oral
tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene
regulation in sickle cell disease: A randomized phase 1 study. PLoS
Med. 2017;14(9):e1002382. https://doi.org/10.1371/journal.pmed.1002382 PMid:28880867 PMCid:PMC5589090
- Lavelle
D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, et al. Effects of
tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral
decitabine. Blood. 2012;119(5):1240-7. https://doi.org/10.1182/blood-2011-08-371690 PMid:22160381 PMCid:PMC3277356
- Yogen
Saunthararajah. Study of Decitabine and Tetrahydrouridine (THU) in
Patients With Sickle Cell Disease (NCT01685515) ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT01685515 (Accessed on November 18, 2019)].
- Meiler
SE, Wade M, Kutlar F, Yerigenahally SD, Xue Y, Moutouh-de Parseval LA,
et al. Pomalidomide augments fetal hemoglobin production without the
myelosuppressive effects of hydroxyurea in transgenic sickle cell mice.
Blood. 2011;118(4):1109-12. https://doi.org/10.1182/blood-2010-11-319137 PMid:21536862 PMCid:PMC3148160
- Celgene.
Study to Determine the Maximum Tolerated Dose, Safety and Effectiveness
of Pomalidomide for Patients With Sickle Cell Disease (SCD-001)
[NCT01522547] ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01522547 (Accessed on November 18, 2019)].
- Srinivas
NR. Clinical pharmacokinetics of panobinostat, a novel histone
deacetylase (HDAC) inhibitor: review and perspectives. Xenobiotica; the
fate of foreign compounds in biological systems. 2017;47(4):354-68.
- Kutlar
A. Study of Panobinostat (LBH589) in Patients With Sickle Cell Disease
(LBH589) [NCT01245179] Clinical Trials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01245179 (Accessed on November 18, 2019)].
- Elias
DB, Barbosa MC, Rocha LB, Dutra LL, Silva HF, Martins AM, et al.
L-arginine as an adjuvant drug in the treatment of sickle cell anaemia.
Br J Haematol. 2013;160(3):410-2. https://doi.org/10.1111/bjh.12114 PMid:23157285
- Chang
J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse
acute vaso-occlusive crises in sickle cell mice through rapid
inhibition of neutrophil adhesion. Blood. 2008;111(2):915-23. https://doi.org/10.1182/blood-2007-04-084061 PMid:17932253 PMCid:PMC2200843
- Albert
Einstein College of Medicine. Intravenous Gammaglobulin for Sickle Cell
Pain Crises (NCT01757418) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01757418 (Accessed on November 18, 2019)].
- Qari
MH, Aljaouni SK, Alardawi MS, Fatani H, Alsayes FM, Zografos P, et al.
Reduction of painful vaso-occlusive crisis of sickle cell anaemia by
tinzaparin in a double-blind randomized trial. Thromb Haemost.
2007;98(2):392-6. https://doi.org/10.1160/Th06-12-0718 PMid:17721622
- Kutlar
A, Ataga KI, McMahon L, Howard J, Galacteros F, Hagar W, et al. A
potent oral P-selectin blocking agent improves microcirculatory blood
flow and a marker of endothelial cell injury in patients with sickle
cell disease. Am J Hematol. 2012;87(5):536-9. https://doi.org/10.1002/ajh.23147 PMid:22488107
- Duke
University. Treatment of Sickle Cell Patients Hospitalized in Pain
Crisis With Prophylactic Dose Low-molecular-weight Heparin (LMWH)
Versus Placebo (NCT01419977) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01419977 (Accessed on November 18, 2019)].
- Novartis
Pharmaceuticals. Study of Two Doses of Crizanlizumab Versus Placebo in
Adolescent and Adult Sickle Cell Disease Patients (STAND) [NCT03814746]
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03814746 (Accessed on November 21, 2019)].
- Novartis
Pharmaceuticals. A Study to Evaluate the Safety and Efficacy of
Crizanlizumab in Sickle Cell Disease Related Priapism (SPARTAN)
[NCT03938454] ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03938454 (Accessed on November 21, 2019)].
- Novartis
Pharmaceuticals. Study of Dose Confirmation and Safety of Crizanlizumab
in Pediatric Sickle Cell Disease Patients (NCT03474965)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03474965 (Accessed on November 21, 2019)].
- Novartis
Pharmaceuticals. Pharmacokinetics and Pharmacodynamics Study of SEG101
(Crizanlizumab) in Sickle Cell Disease (SCD) Patients With Vaso-
Occlusive Crisis (VOC) [NCT03264989] ClinicalTrials.gov [Available
from: https://clinicaltrials.gov/ct2/show/NCT03264989 (Accessed on November 21, 2019)].
- De
Castro LM, Zennadi R, Jonassaint JC, Batchvarova M, Telen MJ. Effect of
propranolol as antiadhesive therapy in sickle cell disease. Clin Transl
Sci. 2012;5(6):437-44. https://doi.org/10.1111/cts.12005 PMid:23253664 PMCid:PMC3762678
- DeCastro
LM. Study of Propranolol as Anti-Adhesive Therapy in Sickle Cell
Disease (SCD) [NCT01077921] ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01077921 (Accessed on November 18, 2019)].
- Field
JJ, Lin G, Okam MM, Majerus E, Keefer J, Onyekwere O, et al. Sickle
cell vaso-occlusion causes activation of iNKT cells that is decreased
by the adenosine A2A receptor agonist regadenoson. Blood.
2013;121(17):3329-34. https://doi.org/10.1182/blood-2012-11-465963 PMid:23377438 PMCid:PMC3637009
- Field
JJ, Majerus E, Gordeuk VR, Gowhari M, Hoppe C, Heeney MM, et al.
Randomized phase 2 trial of regadenoson for treatment of acute
vaso-occlusive crises in sickle cell disease. Blood Adv.
2017;1(20):1645-9. https://doi.org/10.1182/bloodadvances.2017009613 PMid:29296811 PMCid:PMC5728341
- Scheuplein
F, Thariath A, Macdonald S, Truneh A, Mashal R, Schaub R. A humanized
monoclonal antibody specific for invariant Natural Killer T (iNKT)
cells for in vivo depletion. PLoS One. 2013;8(9):e76692. https://doi.org/10.1371/journal.pone.0076692 PMid:24086759 PMCid:PMC3785425
- Field
JJ, Majerus E, Ataga KI, Vichinsky EP, Schaub R, Mashal R, et al.
NNKTT120, an anti-iNKT cell monoclonal antibody, produces rapid and
sustained iNKT cell depletion in adults with sickle cell disease. PLoS
One. 2017;12(2):e0171067. https://doi.org/10.1371/journal.pone.0171067 PMid:28152086 PMCid:PMC5289534
- Hoppe
C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study
of the short-term use of simvastatin in sickle cell disease: effects on
markers of vascular dysfunction. Br J Haematol. 2011;153(5):655-63. https://doi.org/10.1111/j.1365-2141.2010.08480.x PMid:21477202 PMCid:PMC3601917
- University
of North Carolina Chapel Hill. Effect of Atorvastatin on Endothelial
Dysfunction and Albuminuria in Sickle Cell Disease (ENDO) [NCT01732718]
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01732718 (Accessed on November 18, 2019)].
- Children's
Hospital Medical Center Cincinnati. Trial of Zileuton CR in Children
and Adults with Sickle Cell Disease ClinicalTrials.gov [Available from:
http://clinicaltrials.gov/ct2/show/NCT01136941?term=nct01136941&rank=1 (Accessed on November 18, 2019)].
- Zafarullah
M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of
N-acetylcysteine actions. Cell Mol Life Sci. 2003;60(1):6-20. https://doi.org/10.1007/s000180300001 PMid:12613655
- Pace
BS, Shartava A, Pack-Mabien A, Mulekar M, Ardia A, Goodman SR. Effects
of N-acetylcysteine on dense cell formation in sickle cell disease. Am
J Hematol. 2003;73(1):26-32. https://doi.org/10.1002/ajh.10321 PMid:12701116
- Academisch
Medisch Centrum - Universiteit van Amsterdam (AMC-UvA).
N-Acetylcysteine in Patients With Sickle Cell Disease (NAC)
[NCT01849016] ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01849016 (Accessed on November 18, 2019)].
- Novartis Pharmaceuticals. ILARIS (canakinumab) [Package insert]. East Hanover, NJ.2012.
- Novartis
Pharmaceuticals. Study of Efficacy, Safety and Tolerability of ACZ885
(Canakinumab) in Pediatric and Young Adult Patients With Sickle Cell
Anemia (NCT02961218) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02961218 (Accessed on December 16, 2019)].
- Rees
DC, Kilinc Y, Unal S, Dampier C, Pace BS, Kaya B, et al. Double-Blind,
Randomized Study of Canakinumab Treatment in Pediatric and Young Adult
Patients with Sickle Cell Anemia. Blood. 2019;134 (Suppl_1):615.
- Gilead Sciences Inc. Letairis (ambrisentan) tablets [Package insert]. Foster City, CA.2015.
- Augusta University. The Role of Endothelin-1 in Sickle Cell Disease (NCT02712346) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02712346 (Accessed on December 16, 2019)].
- Kutlar
A, Pollock J, Meiler SE, Harris R, Hongyan X, Wells L, et al. Phase-I
Study of ETA Receptor Antagonist Ambrisentan in Sickle Cell Disease.
Blood. 2019;134 (Suppl_1):617.
- Lal A,
Atamna W, Killilea DW, Suh JH, Ames BN. Lipoic acid and
acetyl-carnitine reverse iron-induced oxidative stress in human
fibroblasts. Redox Rep. 2008;13(1):2-10. https://doi.org/10.1179/135100008X259150 PMid:18284845
- UCSF
Benioff Children's Hospital Oakland. Antioxidant Therapy to Reduce
Inflammation in Sickle Cell Disease (NCT01054768) ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT01054768 (Accessed on November 18, 2019)].
- Martins
VD, Manfredini V, Peralba MC, Benfato MS. Alpha-lipoic acid modifies
oxidative stress parameters in sickle cell trait subjects and sickle
cell patients. Clin Nutr. 2009;28(2):192-7. https://doi.org/10.1016/j.clnu.2009.01.017 PMid:19231043
- Christen
JR, Bertolino J, Jean E, Camoin L, Ebbo M, Harle JR, et al. Use of
Direct Oral Anticoagulants in Patients with Sickle Cell Disease and
Venous Thromboembolism: A Prospective Cohort Study of 12 Patients.
Hemoglobin. 2019:1-4. https://doi.org/10.1080/03630269.2019.1689997 PMid:31724442
- Morris
CR, Morris SM, Jr., Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart
D, et al. Arginine therapy: a new treatment for pulmonary hypertension
in sickle cell disease? Am J Respir Crit Care Med. 2003;168(1):63-9. https://doi.org/10.1164/rccm.200208-967OC PMid:12626350
- Morris
CR, Ansari M, Lavrisha L, et al. Arginine therapy for vaso-occlusive
pain episodes in sickle cell disease. Blood (ASH Annual Meeting
Abstracts). 2009;114:573. https://doi.org/10.1182/blood.V114.22.573.573
- Morris
CR, Kuypers FA, Lavrisha L, Ansari M, Sweeters N, Stewart M, et al. A
randomized, placebo-controlled trial of arginine therapy for the
treatment of children with sickle cell disease hospitalized with
vaso-occlusive pain episodes. Haematologica. 2013;98(9):1375-82. https://doi.org/10.3324/haematol.2013.086637 PMid:23645695 PMCid:PMC3762093
- UCSF
Benioff Children's Hospital Oakland. Effectiveness of Arginine as a
Treatment for Sickle Cell Anemia (Arginine) [NCT00513617]
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00513617 (Accessed on November 18, 2019)].
- Hospital de Clinicas de Porto Alegre. L-Arginine and Sickle Cell Disease (NCT01142219) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01142219 (Accessed on November 18, 2019)].
- National
Institutes of Health Clinical Center. Evaluation of Hydroxyurea Plus
L-arginine or Sildenafil to Treat Sickle Cell Anemia (NCT00056433)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00056433 (Accessed on November 18, 2019)].
- UCSF
Benioff Children's Hospital Oakland. Arginine Treatment of Acute Chest
Syndrome (Pneumonia) in Sickle Cell Disease Patients (NCT00029731)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00029731 (Accessed on November 18, 2019)].
- Perrine
SP. Phase II Randomized Trial:Arginine Butyrate Plus Standard Local
Therapy in Patients With Refractory Sickle Cell Ulcers (NCT00004412)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00004412 (Accessed on November 18, 2019)].
- Gladwin
MT, Kato GJ, Weiner D, Onyekwere OC, Dampier C, Hsu L, et al. Nitric
oxide for inhalation in the acute treatment of sickle cell pain crisis:
a randomized controlled trial. JAMA. 2011;305(9):893-902. https://doi.org/10.1001/jama.2011.235 PMid:21364138 PMCid:PMC3403835
- Lopez
BL, Davis-Moon L, Ballas SK, Ma XL. Sequential nitric oxide
measurements during the emergency department treatment of acute
vasoocclusive sickle cell crisis. Am J Hematol. 2000;64(1):15-9. https://doi.org/10.1002/(SICI)1096-8652(200005)64:1<15::AID-AJH3>3.0.CO;2-P
- Head
CA, Swerdlow P, McDade WA, Joshi RM, Ikuta T, Cooper ML, et al.
Beneficial effects of nitric oxide breathing in adult patients with
sickle cell crisis. Am J Hematol. 2010;85(10):800-2. https://doi.org/10.1002/ajh.21832 PMid:20799359
- Ikuta
T, Thatte HS, Tang JX, Mukerji I, Knee K, Bridges KR, et al. Nitric
oxide reduces sickle hemoglobin polymerization: potential role of
nitric oxide-induced charge alteration in depolymerization. Arch
Biochem Biophys. 2011;510(1):53-61. https://doi.org/10.1016/j.abb.2011.03.013 PMid:21457702 PMCid:PMC3889650
- Pfizer.
Safety, Tolerability, Pharmacokinetics, And Pharmacodynamics Study Of
PF-04447943, Co-Administered With And Without Hydroxyurea, In Subjects
With Stable Sickle Cell Disease (NCT02114203) ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT02114203 (Accessed on November 18, 2019)].
- Charnigo
RJ, Beidler D, Rybin D, Pittman DD, Tan B, Howard J, et al.
PF-04447943, a Phosphodiesterase 9A Inhibitor, in Stable Sickle Cell
Disease Patients: A Phase Ib Randomized, Placebo-Controlled Study. Clin
Transl Sci. 2019;12(2):180-8. https://doi.org/10.1111/cts.12604 PMid:30597771 PMCid:PMC6440678
- Singh N, Patra S. Phosphodiesterase 9: insights from protein structure and role in therapeutics. Life Sci. 2014;106(1-2):1-11. https://doi.org/10.1016/j.lfs.2014.04.007 PMid:24746902
- Imara
Inc. An Extension Study of IMR-687 in Adult Patients With Sickle Cell
Anemia (NCT04053803) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT04053803 (Accessed on November 22, 2019)].
- Kato
GJ. A Multi-Center Study of Riociguat in Patients With Sickle Cell
Diseases (NCT02633397) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02633397 (Accessed on November 22, 2019)].
- Cyclerion
Therpeutics. A Study of the Effect of IW-1701 (Olinciguat), a
Stimulator of Soluble Guanylate Cyclase (sGC), on Patients With Sickle
Cell Disease (SCD) (STRONG SCD) [NCT03285178] ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT03285178 (Accessed on November 22, 2019)].
- Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017;129(20):2719-26. https://doi.org/10.1182/blood-2017-02-765891 PMid:28385699 PMCid:PMC5437829
- Geng
X, Dufu K, Hutchaleelaha A, Xu Q, Li Z, Li CM, et al. Increased
hemoglobin-oxygen affinity ameliorates bleomycin-induced hypoxemia and
pulmonary fibrosis. Physiological reports. 2016;4(17). https://doi.org/10.14814/phy2.12965 PMid:27624688 PMCid:PMC5027366
- Howard
J, Hemmaway CJ, Telfer P, Layton DM, Porter J, Awogbade M, et al. A
phase 1/2 ascending dose study and open-label extension study of
voxelotor in patients with sickle cell disease. Blood.
2019;133(17):1865-75. https://doi.org/10.1182/blood-2018-08-868893 PMid:30655275 PMCid:PMC6484388
- Forma
Therapeutics Inc. A SAD/MAD to Assess the Safety, Pharmacokinetics and
Pharmacodynamics of FT-4202 in Healthy Volunteers and Sickle Cell
Disease Patients (NCT03815695) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03815695 (Accessed on December 16, 2019)].
- Kalfa
TA, Kuypers FA, Telen MJ, Malik P, Konstantinidis DG, Estepp JH, et al.
Phase 1 Single (SAD) and Multiple Ascending Dose (MAD) Studies of the
Safety, Tolerability, Pharmacokinetics (PK) and Pharmacodynamics (PD)
of FT-4202, an Allosteric Activator of Pyruvate Kinase-R, in Healthy
and Sickle Cell Disease Subjects. Blood. 2019;134 (Suppl_1):616.
- National
Heart Lung and Blood Institute (NHLBI). Niacin to Improve Blood Flow in
People With Sickle Cell Disease (NCT00508989) ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT00508989 (Accessed on November 18, 2019)].
- Icahn
School of Medicine at Mount Sinai. Vitamin D3 in Patients With Sickle
Cell Disease (NCT03012555) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT03012555 (Accessed on November 18, 2019)].
- Soe
HH, Abas AB, Than NN, Ni H, Singh J, Said AR, et al. Vitamin D
supplementation for sickle cell disease. Cochrane Database Syst Rev.
2017;1:Cd010858. https://doi.org/10.1002/14651858.CD010858.pub2 PMCid:PMC6464759
References
- Greenberg J, Ohene-Frempong K, Halus J, Way C,
Schwartz E. Trial of low doses of aspirin as prophylaxis in sickle cell
disease. J Pediatr. 1983;102(5):781-4. https://doi.org/10.1016/S0022-3476(83)80258-3
- Baxalta
now part of Shire. A Single Dose Study of the Safety, Blood Levels and
Biological Effects of Aes-103 Compared to Placebo in Subjects With
Stable Sickle Cell Disease (NCT01597401) ClinicalTrials.gov [Available
from: https://clinicaltrials.gov/ct2/show/NCT01597401 (Accessed on November 18, 2019).
- Xu
GG, Pagare PP, Ghatge MS, Safo RP, Gazi A, Chen Q, et al. Design,
Synthesis, and Biological Evaluation of Ester and Ether Derivatives of
Antisickling Agent 5-HMF for the Treatment of Sickle Cell Disease. Mol
Pharm. 2017;14(10):3499-511. https://doi.org/10.1021/acs.molpharmaceut.7b00553 PMid:28858508 PMCid:PMC5871537
- Joiner
CH, Jiang M, Claussen WJ, Roszell NJ, Yasin Z, Franco RS. Dipyridamole
inhibits sickling-induced cation fluxes in sickle red blood cells.
Blood. 2001;97(12):3976-83. https://doi.org/10.1182/blood.V97.12.3976 PMid:11389043
- Desai
PC, Brittain JE, Jones SK, McDonald A, Wilson DR, Dominik R, et al. A
pilot study of eptifibatide for treatment of acute pain episodes in
sickle cell disease. Thromb Res. 2013;132(3):341-5. https://doi.org/10.1016/j.thromres.2013.08.002 PMid:23973010 PMCid:PMC3791139
- HemaQuest
Pharmaceuticals Inc. A Study of HQK-1001 in Patients With Sickle Cell
Disease (NCT01322269) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01322269 (Accessed on November 18, 2019).
- Gladwin
MT, Kato GJ, Weiner D, Onyekwere OC, Dampier C, Hsu L, et al. Nitric
oxide for inhalation in the acute treatment of sickle cell pain crisis:
a randomized controlled trial. JAMA. 2011;305(9):893-902. https://doi.org/10.1001/jama.2011.235 PMid:21364138 PMCid:PMC3403835
- Waugh
WH, Daeschner CW, 3rd, Files BA, McConnell ME, Strandjord SE. Oral
citrulline as arginine precursor may be beneficial in sickle cell
disease: early phase two results. J Natl Med Assoc. 2001;93(10):363-71.
- Brousseau
DC, Scott JP, Badaki-Makun O, Darbari DS, Chumpitazi CE, Airewele GE,
et al. A multicenter randomized controlled trial of intravenous
magnesium for sickle cell pain crisis in children. Blood.
2015;126(14):1651-7. https://doi.org/10.1182/blood-2015-05-647107 PMid:26232172 PMCid:PMC4591790
- Belcher
JD, Young M, Chen C, Nguyen J, Burhop K, Tran P, et al. MP4CO, a
pegylated hemoglobin saturated with carbon monoxide, is a modulator of
HO-1, inflammation, and vaso-occlusion in transgenic sickle mice.
Blood. 2013;122(15):2757-64. https://doi.org/10.1182/blood-2013-02-486282 PMid:23908468 PMCid:PMC4067504
- Sangart. Phase 2 Study of MP4CO to Treat Vaso-occlusive Sickle Crisis (NCT01925001) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01925001 (Accessed on November 18, 2019).
- Orringer
EP, Casella JF, Ataga KI, Koshy M, Adams-Graves P, Luchtman-Jones L, et
al. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis
of sickle cell disease: A randomized controlled trial. JAMA.
2001;286(17):2099-106. https://doi.org/10.1001/jama.286.17.2099 PMid:11694150
- Ballas
SK, Files B, Luchtman-Jones L, Benjamin L, Swerdlow P, Hilliard L, et
al. Safety of purified poloxamer 188 in sickle cell disease: phase I
study of a non-ionic surfactant in the management of acute chest
syndrome. Hemoglobin. 2004;28(2):85-102. https://doi.org/10.1081/HEM-120035919 PMid:15182051
- Mast
Therapeutics Inc. Phase III Randomized Study of Poloxamer 188 for
Vaso-Occlusive Crisis of Sickle Cell Disease (NCT00004408)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00004408 (Accessed on November 18, 2019).
- Daak
AA, Ghebremeskel K, Hassan Z, Attallah B, Azan HH, Elbashir MI, et al.
Effect of omega-3 (n-3) fatty acid supplementation in patients with
sickle cell anemia: randomized, double-blind, placebo-controlled trial.
Am J Clin Nutr. 2013;97(1):37-44. https://doi.org/10.3945/ajcn.112.036319 PMid:23193009
- Miller RE. Omega-3 Fatty Acids in Sickle Cell Disease (NCT02947100) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02947100 (Accessed on November 18, 2019).
- Heeney
MM, Hoppe CC, Abboud MR, Inusa B, Kanter J, Ogutu B, et al. A
Multinational Trial of Prasugrel for Sickle Cell Vaso-Occlusive Events.
N Engl J Med. 2016;374(7):625-35. https://doi.org/10.1056/NEJMoa1512021 PMid:26644172
- Ataga
KI, Reid M, Ballas SK, Yasin Z, Bigelow C, James LS, et al.
Improvements in haemolysis and indicators of erythrocyte survival do
not correlate with acute vaso-occlusive crises in patients with sickle
cell disease: a phase III randomized, placebo-controlled, double-blind
study of the Gardos channel blocker senicapoc (ICA-17043). Br J
Haematol. 2011;153(1):92-104. https://doi.org/10.1111/j.1365-2141.2010.08520.x PMid:21323872
- Icagen.
A Study Evaluating the Long-Term Safety of ICA-17043 in Sickle Cell
Disease Patients With or Without Hydroxyurea Therapy (NCT00294541)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00294541 (Accessed on November 18, 2019).
- Machado
RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, et al.
Sildenafil therapy in patients with sickle cell disease and pulmonary
hypertension. Br J Haematol. 2005;130(3):445-53. https://doi.org/10.1111/j.1365-2141.2005.05625.x PMid:16042696 PMCid:PMC2063570
- Machado
RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, et al.
Hospitalization for pain in patients with sickle cell disease treated
with sildenafil for elevated TRV and low exercise capacity. Blood.
2011;118(4):855-64. https://doi.org/10.1182/blood-2010-09-306167 PMid:21527519 PMCid:PMC3148167
- Montefiore
Medical Center. Topical Sodium Nitrite in Sickle Cell Disease and Leg
Ulcers (NCT02863068) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02863068 (Accessed on November 18, 2019).
- TRF Pharma Inc. TRF-1101 Assessment in Sickle Cell Disease (NCT00773890) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT00773890 (Accessed on November 18, 2019).
- Anthera
Pharmaceuticals. A Study of Varespladib Infusion in Subjects With
Sickle Cell Disease. (IMPACTS-2) [NCT01522196] ClinicalTrials.gov
[Available from: https://clinicaltrials.gov/ct2/show/NCT01522196 (Accessed on November 18, 2019).
- Mast
Therapeutics Inc. Evaluation of Purified Poloxamer 188 in
Vaso-Occlusive Crisis of Sickle Cell Disease (EPIC) [NCT01737814]
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT01737814?term=purified+poloxamer+188&rank=1 (Accessed on November 18, 2019).
- Dana-Faber
Cancer Institute. Efficacy of Vorinostat to Induce Fetal Hemoglobin in
Sickle Cell Disease (NCT01000155) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT0100015 5 (Accessed on November 18, 2019).
- Modus
Therapeutics AB. Sevuparin Infusion for the Management of Acute VOC in
Subjects With SCD (NCT02515838) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/record/NCT02515838 (Accessed on November 18, 2019).
- Kutlar A, Embury SH. Cellular adhesion and the endothelium: P-selectin. Hematol Oncol Clin North Am. 2014;28(2):323-39. https://doi.org/10.1016/j.hoc.2013.11.007 PMid:24589269
- Pfizer.
Efficacy and Safety of Rivipansel (GMI-1070) in the Treatment of
Vaso-Occlusive Crisis in Hospitalized Subjects With Sickle Cell Disease
(NCT02187003) ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02187003 (Accessed on November 18, 2019).
- Prolong
Pharmaceuticals. Study of SANGUINATE™ In the Treatment of Sickle Cell
Disease Patients With Vaso-Occlusive Crisis (NCT02411708)
ClinicalTrials.gov [Available from: https://clinicaltrials.gov/ct2/show/NCT02411708 (Accessed on November 18, 2019).
[TOP]