George J. Kontoghiorghes, Marios Kleanthous and Christina N. Kontoghiorghe.
Postgraduate Research Institute of Science, Technology, Environment and Medicine, Limassol, Cyprus.
Corresponding
author:George J. Kontoghiorghes,
Postgraduate Research Institute of Science, Technology, Environment and
Medicine, 3 Ammochostou Street, Limassol 3021, Cyprus. Tel:
+35726272076; Fax: +35726272076. E-mail:
kontoghiorghes.g.j@pri.ac.cy
Published: January 1, 2019
Received: October 29, 2019
Accepted: December 18, 2019
Mediterr J Hematol Infect Dis 2020, 12(1): e2020011 DOI
10.4084/MJHID.2020.011
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Deferiprone
(L1) was originally designed, synthesised and screened in vitro and in
vivo in 1981 by Kontoghiorghes G. J. following his discovery of the
novel alpha-ketohydroxypyridine class of iron chelators (1978-1981),
which were intended for clinical use. The journey through the years for
the treatment of thalassaemia with L1 has been a very difficult one
with an intriguing turn of events, which continue until today. Despite
many complications, such as the extensive use of L1 suboptimal dose
protocols, the aim of chelation therapy- namely, the complete removal
of excess iron in thalassaemia major patients, has been achieved in
most cases following the introduction of specific L1 and
L1/deferoxamine combinations. Many such patients continue to maintain
normal iron stores. Thalassemia has changed from a fatal to chronic
disease; also thanks to L1 therapy and thalassaemia patients are active
professional members in all sectors of society, have their own families
with children and grandchildren and their lifespan is approaching that
of normal individuals. No changes in the low toxicity profile of L1
have been observed in more than 30 years of clinical use and
prophylaxis against the low incidence of agranulocytosis is maintained
using mandatory monitoring of weekly white blood cells’ count.
Thousands of thalassaemia patients are still denied the
cardioprotective and other beneficial effects of L1 therapy. The safety
of L1 in thalassaemia and other non-iron loaded diseases resulted in
its selection as one of the leading therapeutics for the treatment of
Friedreich’s ataxia, pantothenate kinase-associated neurodegeneration
and other similar cases. There are also increasing prospects for the
application of L1 as a main, alternative or adjuvant therapy in many
pathological conditions including cancer, infectious diseases and as a
general antioxidant for diseases related to free radical pathology.
|
Introduction
The
haemoglobinopathies, which include sickle cell disease and
thalassaemia, are a major group of genetic diseases affecting humans.
It is estimated that about 100000 children are born annually with
thalassaemia mainly in South-East Asia, the Middle East and
Mediterranean countries. Most of the thalassaemia patients are born in
South-East Asia and die untreated.[1,2]
Thalassaemia
is endemic in some countries such as Cyprus, where 1 in 6 persons is an
asymptomatic thalassaemia heterozygote carrier, and about 1 in 1000 is
a thalassaemia major and intermedia patient. The prevention and
treatment programmes for thalassaemia and especially chelation therapy
impose a great financial burden on the health budget of many countries.[1,3]
The
standard form of treatment of transfusion dependent thalassaemia (TDT)
is regular red blood cell (RBC) transfusions every 1-4 weeks
accompanied by daily chelation therapy. Multiple transfusions cause
increased iron deposition and damage to the liver, heart, spleen and
other organs. Iron overload in thalassaemia has the highest rate of
morbidity and mortality of metal related intoxication in humans. Iron
chelation therapy in thalassaemia and other transfusional iron loading
conditions is carried out worldwide using the generic drugs
deferoxamine (DF), deferiprone (L1) and deferasirox (DFRA).[4]
The combination of chelating drugs and especially the L1/DF
combination, is also widely used in the vast majority of thalassaemia
patients in Cyprus and also in some other countries.[5,6]
There
is no worldwide consensus in the use of iron chelating drugs or of
related protocols in transfusional iron loaded patients. In most cases,
the selection of chelation therapy and related protocols is generally
‘random’ and based on subjective and other criteria and non specific
aim. As a result, the selection and use of iron chelating drugs vary
from country to country and from clinic to clinic.[7]
The
main aim of iron chelation therapy in thalassaemia and other iron
loaded conditions is the achievement and maintenance of normal iron
stores, in which case patients are devoid of iron overload toxic side
effects.[8] This aim, including the long term
prevention of iron overload, can be accomplished using effective and
safe chelation protocols, involving mainly L1 and L1/DF combinations.[8]
Some
of the unique pharmacological properties and characteristics of L1 such
as the ability to penetrate most organs and remove effectively excess
iron from the heart has resulted in a substantial reduction in the
mortality rate of thalassaemia.[4-6,9,10]
Furthermore, the ability of L1 to remove excess iron from the brain has
resulted in the development of L1 as one of the leading pharmaceuticals
in the treatment of Friedreich’s ataxia, pantothenate kinase-associated
neurodegeneration (PKAN), and other cases of neurodegeneration with
brain iron accumulation.[11-13]
Design, Development, and Cost of Deferiprone
The
design, development, and clinical use of L1 is a unique case of
academic orphan drug development, which was originally based on
academic efforts supported mainly by a thalassaemia patient/parent
charitable organisation, namely the United Kingdom Thalassaemia Society
(UKTS).
The project on chelation was initiated at the University
of Essex, UK, while working on haemoglobins in 1979 and was partly
supported by the British Technology Group (BTG) and the UKTS.[14,15]
Following
a fundamental new approach on iron chelation design and testing, a new
group of iron chelators was discovered by one of the authors- namely
Kontoghiorghes G J (GJK), and as a result, the new classes of
alpha-ketohydroxyridines including 1,2-dimethyl-3-hydroxypyrid-4-one
(L1) were synthesised and tested at Essex University and University
College Medical School London (UCH), UK.[15,16] The latter was selected by GJK and members of UKTS because of the in vivo and clinical testing facilities.[15-18]
The
discovery and iron removal effects of L1 could not be published for 5
years due to “embargo” on publications by BTG, and in 1985 the UKTS
sponsored the continuation of the chelation project at the Royal Free
Hospital Medical School (RF) London, UK from where the first
publications of the iron removal effects of L1 in comparison to
parenteral DF in animals were reported.[19-22]
A
significant invention at the RF in 1986 was also the one-step novel
synthesis of L1 and L1 analogues, instead of the 4-step synthesis
invented in 1981, which overturned the BTG patent monopoly in many
countries. The one-step synthetic method is currently utilised by all
manufacturers of L1 worldwide and make L1 less expensive than DF and
DFRA.[23-27] Deferiprone became a generic drug about
15 years ago, and by comparison, its sale price in India, Iran and
Thailand is about 5-10 times cheaper than that sold in western
countries.[23-27]
A fierce competition against
L1 and related controversies were in process from the time of the L1
discovery until today. For example, more than 60 patents were filed
worldwide since the discovery of L1 and other alpha-ketohydroxyridines.[15] The first patent application was filed in 1982.[15,28]
However, due to ‘policy changes’, BTG has submitted the remaining
patents under the names of the inventor (GJK) and co-inventors using an
alphabetical order format.[29,30]
An analogue of
L1, namely 1,2-diethyl-3-hydroxypyrid-4-one (EL1NEt or CP 94) was
promoted by BTG sponsored studies at Essex University and UCH.[31-35] However, based on further studies and clinical trials in thalassaemia patients, EL1NEt was later abandoned.[36-39]
Similarly, Ciba Geigy (now Novartis) the then manufacturers of DF have
also carried out animal toxicity studies with L1 and reached the
conclusion that L1 was toxic.[40] However, the
evaluation methods used for L1, as well as the comparative toxicity
data obtained previously with DF questioned Ciba Geigy’s conclusions.[41] Similar controversies continue until today.[42-45]
Despite
the opposition from different groups, the academic initiative and
strategy for the development of L1 in academic institutions continued
and included the general phase I to V studies as described for most
other drugs.[45]
The First Clinical Trials with Deferiprone and Today’s Implications
Approval
for the first clinical trials with L1 in myelodysplasia and
thalassaemia patients was obtained in 1987 from the local ethical
committee of the RF and the Department of Health and Social Securities
of the UK.[46,47] Gelatine capsules were used for the oral administration of L1 because of its bitter taste (Figure 1).
 |
Figure 1. The first
pharmaceutical preparation of encapsulated deferiprone (L1).
Encapsulated 0.5 g L1 white solid in transparent gelatin capsules used
for the first clinical trials in London, UK and in multicentre clinical
trials that followed. No preservatives or additives were included in
the preparation. This simple formulation masked the bitter taste of L1. |
In
the first two clinical studies, different divided or single daily doses
of L1 were administered to 11 patients (10-110 mg/kg/day) to assess
efficacy and tolerability.[46,47] All doses caused a
net increase of urinary iron excretion (UIE), which was proportional to
the dose of L1 and the iron load of the patients (Figure 2).
Doses of 75-110 mg/kg/day were identified to cause negative iron
balance with an increase in UIE greater than 25-33 mg, which was
equivalent to that caused by DF and higher than the intake of iron from
RBC transfusions.[47] No increase in urinary excretion of other essential metals (Ca, Zn and Mg) or other toxic side effects were reported.[46,47]
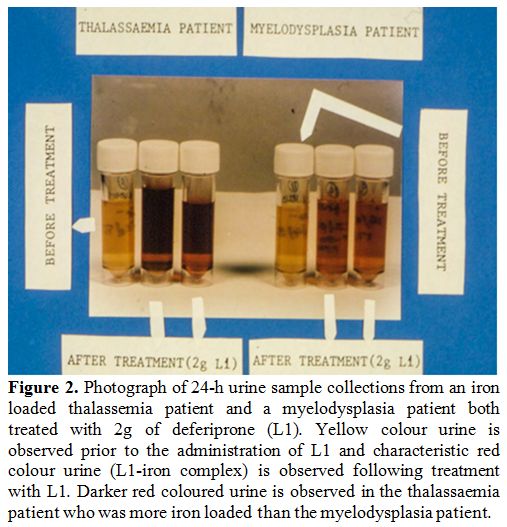 |
Figure 2. Photograph of
24-h urine sample collections from an iron loaded thalassemia patient
and a myelodysplasia patient both treated with 2g of deferiprone (L1).
Yellow colour urine is observed prior to the administration of L1 and
characteristic red colour urine (L1-iron complex) is observed following
treatment with L1. Darker red coloured urine is observed in the
thalassaemia patient who was more iron loaded than the myelodysplasia
patient. |
International
multicentre clinical trials were organised, following the initial
clinical trials in London and L1 was supplied in different university
clinical centres worldwide e.g. Italy, Switzerland, The Netherlands,
Germany, etc.[21] The production of L1 for clinical
trials was later carried out by private companies in India,
Switzerland, The Netherlands and Canada etc.[21]
In
1989 the first episode of agranulocytosis was reported, as well as
episodes of neutropenia, masculoskeletal and joint pains, gastric
intolerance, and zinc deficiency in the RF, which were also confirmed
by other centres.[48-51] In the same year, no similar
agranulocytosis episodes were observed in a total of 125 other patients
who received L1 in 8 other countries, as reported in the first
international conference on oral chelation (1st ICOC) at the RF.[49]
In this context, a mandatory weekly blood count was introduced for
prophylaxis against agranulocytosis and neutropenia similar to the drug
clozapine.[21]
An application proposing a name
for L1 in 1991 by the inventor (GJK), resulted in the adoption by the
World Health Organisation (WHO) of the INN name deferiprone (WHO drug
information list 67, volume 2 of 1992).
There was no interest from major pharmaceutical companies for the commercial development of L1.[5,40]
Within this context, India played a leading role in the pharmaceutical
development of L1. A collaborative project initiated between a parent
of a thalassaemia patient of the Indian pharmaceutical generic company
Cipla with one of us (GJK), led to the pharmaceutical preparation of L1
and also the initiation of clinical trials in India.[21,50]
The first in the world regulatory approval for L1 was in India and L1
became available to Indian thalassaemia patients in 1995 (Table 1).[49,50]
At a later stage, BTG licensed the L1 patents to the generic
pharmaceutical company Apotex, Canada and L1 received regulatory
approval from the EU in 1999 and many other countries worldwide and
also from the USA in 2011.
 |
Table
1. Deferiprone (L1)- the journey across the years. |
No
formal animal or other preclinical toxicology studies were carried out
on L1 by either Cipla or Apotex. The absence of such data put L1 at a
disadvantage as a second line iron chelating drug in comparison to DF
and DFRA. However, animal toxicology data are generally of a similar
level of toxicity for all three drugs and in clinical practice, L1 is
widely used to the same extent as the other two chelating drugs. In
many cases, L1 is regarded as the first line iron chelating drug
because of its unique properties and especially its cardioprotective
effects.[5-10]
With regards to safety, long term
studies, and continuous clinical monitoring involving thousands of
thalassaemia and other categories of patients in the last 30 years have
confirmed the low toxicity of L1.[50,51] The most
severe toxic side effects of L1 still remain the same until today and
are all controllable, manageable, and reversible. These include
reversible agranulocytosis (1% >) and neutropenia (5% >), while
less serious toxic side effects include gastric intolerance,
masculoskeletal and joint pains and zinc deficiency.[50,51]
Toxicity vigilance and prophylactic measures are essential steps for
ensuring the safety of L1 and the other chelators. For example, zinc
supplements are used for prophylaxis for patients on long term
treatment with L1, DF and the L1/DF combination.[21]
Agranulocytosis
is the most severe toxic side effect of L1 and mandatory monitoring of
weekly white blood cell count is an essential prophylactic measure for
its prevention during L1 therapy. Similarly, temporary withdrawal of L1
is necessary during the sore throat and other infections. The cause of
agranulocytosis is still unknown but in almost all the cases, this L1
toxicity was transient and all the patients recovered following
treatment with granulocyte-colony stimulating factor (G-CSF). The time
of recovery of the neutrophil count in patients treated with G-CSF
varies between a few days to 7 weeks.[21,48]
The mechanism of L1 induced agranulocytosis is thought to be related to
an L1-related immune response against white cell progenitors since
re-challenge on the same patients with L1 results in another episode of
agranulocytosis. The patients with this L1 toxic side effect have to
switch to DF or DFRA chelation.
Mechanisms of Chelation and Prevention of Iron Toxicity by Deferiprone
The properties and mechanisms of chelation by L1 and other chelating drugs have been previously reviewed.[4,21,22]
Three molecules of L1 are needed to bind one molecule of iron and to
form a red colour iron complex similar to that shown in the urines of
iron loaded patients in Figure 2.
The small molecular size, neutral charge, and hydrophilicity of L1
allow substantial penetration of almost all tissues including access to
all major organs such as the heart and the brain.[4,21,22]
As a result of the extensive distribution, L1 can act as a universal
antioxidant in all conditions associated with free radical pathology by
inhibiting oxidative stress damage caused by excess labile iron
catalysis of free radical production.[4,21]
The pharmacokinetics, metabolism and route of iron elimination of L1 have also been determined (Figure 3).[21,52-54]
Deferiprone is readily absorbed within minutes from the stomach,
metabolised to a glucuronide conjugate, cleared from the plasma within
6-8 hours, and excreted in the urine in the form of L1 iron complex, L1
glucuronide conjugate and free L1 (Figure 3).[16,21,52-54] No increase in iron excretion was detected in the faeces of iron loaded thalassaemia patients treated with L1 (Figure 4).[53,55]
Iron mobilisation by L1 depends on the iron load of the patients and the dose of L1 (Figure 4).[53]
The increase of UIE in non iron loaded patients is only a few mg, which
by comparison, is a small fraction of what is excreted in iron loaded
patients.[21,53]
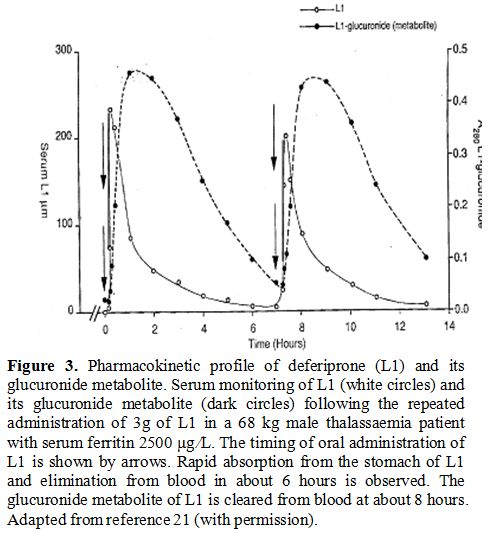 |
Figure 3.
Pharmacokinetic profile of deferiprone (L1) and its glucuronide
metabolite. Serum monitoring of L1 (white circles) and its glucuronide
metabolite (dark circles) following the repeated administration of 3g
of L1 in a 68 kg male thalassaemia patient with serum ferritin 2500 μg
⁄L. The timing of oral administration of L1 is shown by arrows. Rapid
absorption from the stomach of L1 and elimination from blood in about 6
hours is observed. The glucuronide metabolite of L1 is cleared from
blood at about 8 hours. Adapted from reference 21 (with permission). |
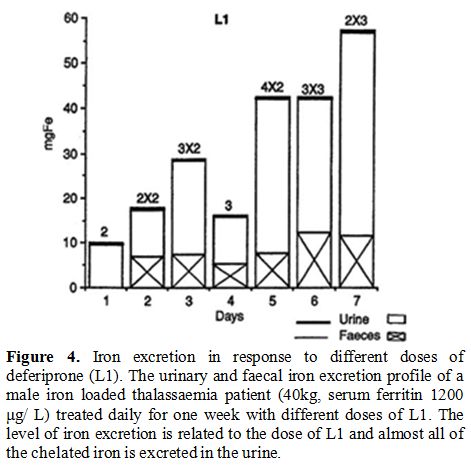 |
Figure 4. Iron excretion in response to
different doses of deferiprone (L1). The urinary and faecal iron
excretion profile of a male iron loaded thalassaemia patient (40kg,
serum ferritin 1200 μg/ L) treated daily for one week with different
doses of L1. The level of iron excretion is related to the dose of L1
and almost all of the chelated iron is excreted in the urine. |
Iron
chelation and mobilisation by L1 have been shown to occur from all the
iron pools in cells including ferritin and haemosiderin and also from
transferrin and NTBI in plasma (Figure 5).[4,21,22,56]
In contrast to the other chelating drugs, only L1 can cause the
mobilisation of iron from transferrin and prevent the accumulation of
excess iron in cells (Figure 5).[4,21,22,56]
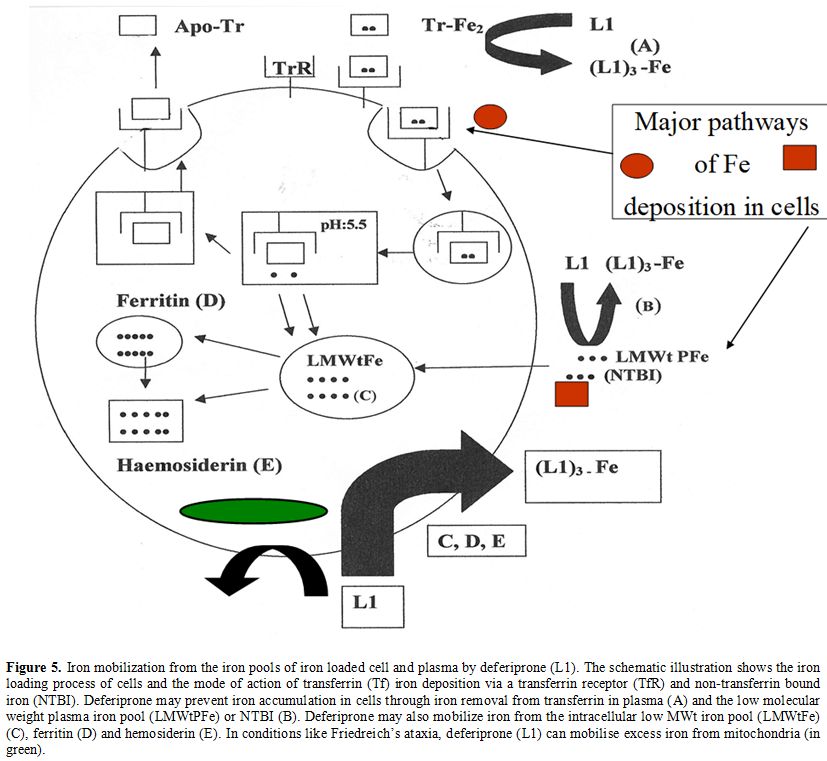 |
Figure 5. Iron
mobilization from the iron pools of iron loaded cell and plasma by
deferiprone (L1). The schematic illustration shows the iron loading
process of cells and the mode of action of transferrin (Tf) iron
deposition via a transferrin receptor (TfR) and non-transferrin bound
iron (NTBI). Deferiprone may prevent iron accumulation in cells through
iron removal from transferrin in plasma (A) and the low molecular
weight plasma iron pool (LMWtPFe) or NTBI (B). Deferiprone may also
mobilize iron from the intracellular low MWt iron pool (LMWtFe) (C),
ferritin (D) and hemosiderin (E). In conditions like Friedreich’s
ataxia, deferiprone (L1) can mobilise excess iron from mitochondria (in
green). |
Many
factors can affect the rate of accumulation and deposition of iron in
the organs of transfused iron loaded patients, with the rate of RBC
transfusions being the primary factor (Table 2).
Similarly, many factors can affect the rate of iron removal from
regularly transfused patients with the most important being the
efficacy of the iron chelation protocol (Table 2).
In this context, the selected chelating drugs and dose protocols, as
well as other related effects, can influence the outcome of chelation
therapies (Table 3).[57,88]
 |
Table 2. Factors affecting the iron load and iron removal in transfused patients. |
 |
Table 3. Comparison of the mode of action and effects of chelating drugs. |
There are many
variables in the properties and mode of action of chelating drugs and
the selection of any chelation protocol could have a direct effect on
the mortality and morbidity of thalassaemia patients (Table 3).[5-10,57-60]
Optimum iron chelation therapies in the context of personalised
medicine in thalassaemia patients take into consideration the most
effective and less toxic monotherapy or combination therapy
protocols.[61] In this context, for each patient, the dose protocols
are adjusted with regards to the iron load and the
efficacy/tolerability of the chelation therapy.[61] The
benefits from the selection of a chelation protocol could easily be
assessed by monitoring the levels of the iron load and also organ
function. For example, the removal of excess toxic iron in thalassaemia
patients by L1, and the L1/DF combination is accompanied with
improvement of cardiac function, such as elevation of left ventricular
ejection fraction (LVEF), endothelial function, etc.[9,10,62]
Improvements have also been observed in some other haematological
conditions using L1 but the mechanisms have not yet been fully
clarified.[63-65]
Recent Developments on Iron Chelation Metabolic Pathways
Congestive
cardiac failure due to cardiac iron overload toxicity has been the
primary cause of mortality in iron loaded thalassaemia patients for
many decades.[66,67] Despite that in thalassaemia the
diagnostic tests previously used routinely for estimating iron stores
such as serum ferritin and liver biopsies could generally reflect body
iron stores, neither of these tests could reflect cardiac iron load
levels.[68-70] Furthermore, such information was not
sufficient for selecting appropriate chelation therapy protocols for
effective removal of excess iron from the heart.
However, the
relatively recent routine introduction of new diagnostic techniques
such as Magnetic Resonance Imaging (MRI) T2 and T2* which identify the
level of iron load in the heart, liver, spleen and other organs of
thalassaemia and other iron overloaded patients, has not only increased
our understanding of transfusional and other iron overload metabolic
pathways but also the differential effect of chelating drugs in iron
removal from various organs.[60,69-73]
The
recent diagnostic procedures, and especially MRI T2 and T2* in the
determination of iron deposition in organs, have increased the
prospects of improved chelating drug targeting therapies of iron
overload toxicity, as well as the introduction of personalised
chelation regimens in thalassaemia and other iron overload metabolic
disorders.[73] Furthermore, based on these diagnostic
findings the complete treatment of iron overload by removing all excess
iron safely from the heart, liver and other organs of regularly TDT
patients using L1, the L1/DF or other chelator combinations can
nowadays be precisely monitored (Figure 6).
In addition, the safe long term maintenance of normal iron stores in
thalassaemia patients and prevention of chelating drug toxicity can
also be regularly assessed using monthly monitoring of serum ferritin
levels, as well as yearly or half yearly MRI T2 and T2* measurements.
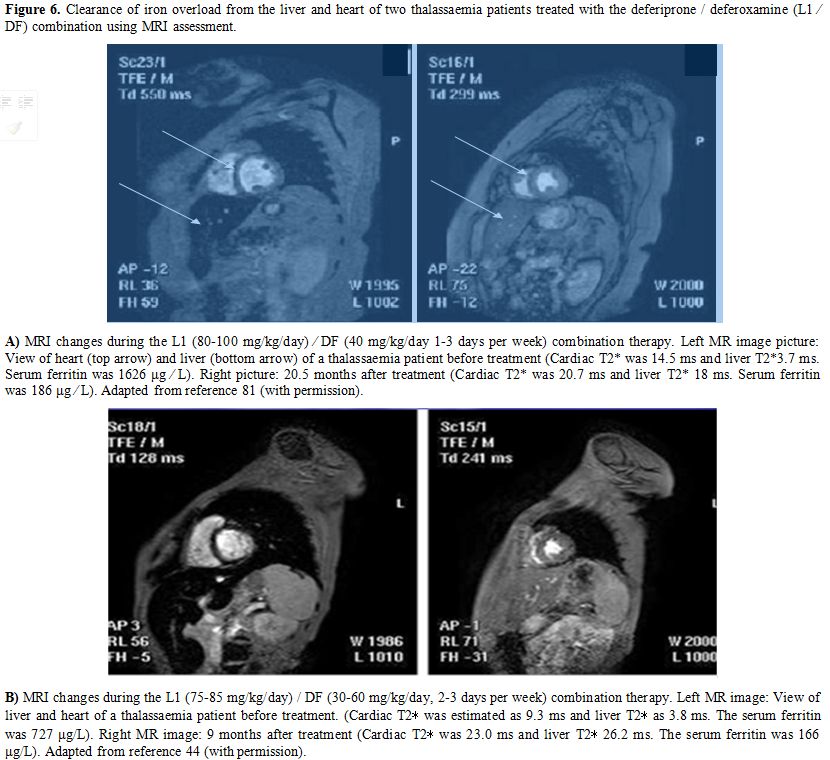 |
Figure 6. Clearance of
iron overload from the liver and heart of two thalassaemia patients
treated with the deferiprone / deferoxamine (L1 ⁄ DF) combination using
MRI assessment. A) MRI changes
during the L1 (80-100 mg/kg/day) ⁄ DF (40 mg/kg/day 1-3 days per week)
combination therapy. Left MR image picture: View of heart (top arrow)
and liver (bottom arrow) of a thalassaemia patient before treatment
(Cardiac T2* was 14.5 ms and liver T2*3.7 ms. Serum ferritin was 1626
μg ⁄ L). Right picture: 20.5 months after treatment (Cardiac T2* was
20.7 ms and liver T2* 18 ms. Serum ferritin was 186 μg ⁄ L). Adapted
from reference 81 (with permission). B)
MRI changes during the L1 (75-85 mg/kg/day) / DF (30-60 mg/kg/day, 2-3
days per week) combination therapy. Left MR image: View of liver and
heart of a thalassaemia patient before treatment. (Cardiac T2∗ was
estimated as 9.3 ms and liver T2∗ as 3.8 ms. The serum ferritin was 727
μg/L). Right MR image: 9 months after treatment (Cardiac T2∗ was 23.0
ms and liver T2∗ 26.2 ms. The serum ferritin was 166 μg/L). Adapted
from reference 44 (with permission).
|
The
promising results in the treatment of iron overload in thalassaemia
encouraged investigations for the use and development of chelating
drugs in many other clinical conditions. Such initiatives were within
the broad context of the risk/benefit assessment of therapeutic
outcomes in each condition because of the absence of other effective
therapeutic approaches. Most efforts were mainly focused on the use of
L1 as a universal antioxidant in non iron overload diseases such as
neurodegenerative, cardiovascular, renal, infectious diseases, as well
as other diseases including cancer and ageing.[74,75]
Recent
developments involving the prospects of the broader use of chelating
drugs have been investigated in clinical trials and clinical
developments in many of these clinical conditions.[74,75]
In particular, the introduction of L1 for the treatment of non iron
loaded patients with focal toxic iron deposits e.g. in Friedreich
ataxia and toxic labile iron e.g. in diabetic and non-diabetic
glomerular disease, is a reflection of the antioxidant and safety
potential of L1.[11-13,74,75] As in
many other cases of drug development, the prospects of introduction of
L1 and other chelating drugs in these diseases are based on commercial
and not ethical criteria.[45]
The Paradigm of the Complete Treatment of Iron Overload in Thalassaemia
The
removal of excess toxic iron accumulated from repeated RBC transfusions
in patients with refractory anaemias was the primary aim of all
investigations involved with iron removal chelation therapy in the last
50 years. In general, any form of excess iron is potentially toxic
because of the ability of iron to catalyse the increased production of
free radicals and cascades, which can cause molecular, subcellular,
cellular, tissue, and organ damage.[15,65]
The extent of damage can be reversible or irreversible depending mainly
on the concentration of excess deposited iron and also other factors (Table 2).[75]
With
the introduction of intramuscular and then subcutaneous and intravenous
DF in the early 1960’s, it became evident that the rate of iron removal
by DF was not sufficient to compensate for the body iron intake from
RBC transfusions in the vast majority of thalassaemia major patients
mainly due to severe complications with the parenteral administration
of DF.[66,67,74] Furthermore,
serious complications were also observed such as neurotoxic and other
toxic side effects during the use of DF in low iron loaded thalassaemia
patients and also other categories of patients with normal iron stores.[66,74]
As
a result of the DF limitations, no clear strategies have become
available or promoted in the last 50 years for the complete elimination
of excess iron and the normalisation of the iron stores in thalassaemia
and other patients.
It has been estimated previously that in the
absence of chelation therapy the mean survival of thalassaemia major
patients was about 20 years, and the primary cause of death was
congestive cardiac failure.[5,66,74]
Results from a UK registry indicate that with the introduction of DF
the mean survival of thalassaemia major patients has increased to about
35 years.[76] Recently, with the introduction of L1,
the mortality rate of thalassaemia major patients has decreased
substantially and mean survival is approaching that of normal
individuals.[5,77]
It appears
that the primary aim of chelation therapy in thalassaemia major and
possibly other chronically transfused patients, i.e. the removal of all
excess iron and inhibition/prevention of iron toxicity, as well as the
associated tissue and organ damage can now be accomplished in most
cases.[8,77] This aim became
foreseeable and applicable very recently especially in patients that
followed the ICOC protocol using L1 and L1/DF combinations.[8,73,77]
Furthermore, the secondary aim of chelation therapy in chronically
transfused patients i.e. the safe maintenance of normal iron stores,
has also been achieved using lower dose ICOC protocols of L1
monotherapy and L1/DF combinations.[8,73,77]
The Achievement and Maintenance of Normal Iron Stores in Thalassaemia
Although
the efficacy of chelation monotherapies with DF, L1, and DFRA have been
thoroughly studied, no normalisation of the iron stores was reported or
investigated in thalassaemia major patients since in the vast majority
of patients the rate of iron removal by chelation was, in general,
lower by comparison to the rate of iron intake from RBC transfusions (Table 2).
The
normalisation of the iron stores in thalassaemia major and other
chronically transfused patients was not considered as a possible option
following the introduction of DF and later DFRA, mainly because DFRA
and DF were not sufficiently effective in removing all excess iron but
also because in both cases there was a high risk of toxicity in non
heavily iron loaded patients with serum ferritin lower than 500 μg/L,
as described in their drug label information.[78]
Another
limiting factor for not achieving normal iron stores was that no such
aim had been proposed in the medical literature until recently or was
described in the drug label information of L1, DF, and DFRA. It appears
that overall insufficiently effective and suboptimal chelating drug
dose protocols are generally used even today by most thalassaemia and
other clinics, despite that the normalisation of the iron stores should
be a primary aim for thalassaemia and other multitransfused patients.
In most of these cases, chelating drug combinations are required for
achieving normal iron stores.[73]
Individual
drug monotherapies are described and recommended by the chelating drug
manufacturers in all three chelating drug label information, while
chelating drug combinations are not described and are clearly excluded
as a form of therapy. The prospect of chelating drug combinations and
precisely the L1/DF combination was an academic initiative and
suggested as early as 1987 and repeated in 1992[46,79] It was then mainly recommended for patients with toxicity or efficacy complications of either DF or L1.[46,79]
The
dilemma of how to control iron load and over-chelation following the
achievement of normal iron stores has been demonstrated in several
studies using the ICOC and similar protocols of tailor-made
administration of L1 and L1/DF combinations and by regular monitoring
of iron store levels.[8,78]
The
first report of the normalisation of the iron stores in iron loaded
thalassaemia major patient was described following the replacement of
DF with L1 due to congestive cardiac failure caused by cardiac iron
overload during DF therapy.[80] Several other reports
followed, indicating that the use of selected combinations of L1 and DF
could achieve the normalisation of the iron stores in iron loaded
thalassaemia major patients.[81-83] In particular,
the ICOC protocol of L1 (80–100 mg/kg/day) and subcutaneous DF (40–60
mg/kg/day, at least 3 days per week) was identified as the most
tolerable and effective chelation therapy protocol for achieving
negative iron balance (Figure 6, Table 3).[33,81]
Continuous
monitoring of the iron stores, e.g. monthly serum ferritin assessment,
is required for regularly transfused patients who have achieved normal
iron stores.[83] Furthermore, continuous adjustment
of iron chelation dose protocols is necessary for maintaining the
normal iron stores without the prospect of excess chelation toxicity.[83]
Different dose protocols of L1, DF, and L1/DF combination, are required
for maintaining normal iron stores within the context of personalised
medicine.[83] In some cases of low serum ferritin in
thalassaemia major patients, withdrawal of chelation therapy may be
necessary for avoiding iron deficiency.[83,84]
In
addition to the more significant clinical benefits for thalassaemia
patients from the maintenance of normal iron stores, there is also a
substantial reduction in the cost of chelation therapy since much lower
doses of chelators are generally used by comparison to iron loaded
paients.[58,83]
Future Prospects of Iron Chelation Therapy
It
is conceivable that the aim of iron chelation therapy in transfusional
iron overload for achieving and maintaining normal iron stores will be
accomplished in many more patients in the forthcoming years, thus
decreasing associated morbidity and mortality due to excess iron
toxicity. Already in countries like Cyprus, many thalassaemia patients
are achieving life spans approaching that of the general population,
are active professionals in society and have families with children and
even grandchildren.[5,77]
The
same aim and approach for the normalisation of the iron stores and the
reduction of excess iron toxicity in thalassaemia major could be used
in many other haematological conditions of iron overload including
myelodysplasia, post-allogenic stem cell trans-plantation,
non-transfusion dependent thalassaemia (NTDT), non-venesected
idiopathic haemochromatosis, transfused cancer cases etc.[78]
Effective iron chelation therapy protocols within the context of
personalised medicine and risk/benefit assessment could be used in each
of these cases, similar to the ICOC protocol.[61] In
most of these cases, tolerant and active combination protocols of 1-3
chelating drugs may be used for effective and rapid clearance of excess
iron.[81-83]
The interaction between chelating
drugs and chelating drugs with other drugs used for other therapeutic
effects of the underlying diseases needs further investigations.
Similarly, the therapeutic and toxic effects of drugs with chelating
potential such as hydroxycarbamate (hydroxyurea) and iron also need
further investigation.[85]
The clinical
application of iron chelating drugs and other chelators is likely to
increase in the future involving the treatment of many other diseases
in addition to transfusional iron overload and focal iron deposit
toxicities.[11-13,74,86] Initial clinical trials in several non iron loaded diseases with L1 are encouraging and promising.[87-90]
Most of these future applications include infectious diseases by
withholding iron from microbes, intervention in iron metabolic pathways
associated with cancer, HIV and other diseases, detoxification of
environmental and diagnostic metals, and inhibition of excess toxic
free radical production involved in many diseases of free radical
pathology.[74,75,87-91] In
particular, with regards to the latter, iron chelation therapy using L1
has been considered for the reduction of anticancer drug toxicity such
as doxorubicin, for ophthalmic toxicity and neurotoxicity and also many
other related applications.[92-95]
The
selection of therapeutic protocols for thalassaemia and other diseases
involving chelating drugs is crucial because it affects risk/benefit
assessment and therapeutic outcome, as well as morbidity and mortality
of hundreds of thousands of patients.[96-99] The
present state of generally ‘random’ selection of chelating drug
protocols does not appear to benefit the patients. In this context, the
high efficacy and safety of the ICOC L1/DF combination protocol should
be considered as a first line chelation treatment for the vast majority
of thalassaemia patients.[8,81,83]
This proposition is supported by recent detailed monitoring findings in
the improvement in cardiac iron depletion rate and cardiac function by
L1 and L1/DF over other therapies.[60,100]
Advances in the constant monitoring of iron deposition in critical
organs like heart, liver, and pancreas by MRI T2* has recently allowed
improvement in the tailoring iron chelation therapy and the selection
of the more appropriate chelation regimens in different clinical cases,
thus reducing overall patient mortality and morbidity.[101-103]
The
limitations in the use of L1 and the L1/DF combination in some
countries may constitute an irregular action by health policy decision
makers and also negligence in relation to the well being of
thalassaemia patients. This policy appears to be controversial,
especially considering that drug combinations are widely used not only
in other haematological conditions but also in many other diseases.
Similar
controversies apply in the risk/benefit assessment for the use of
chelating drugs not only in transfusion-dependent thalassemia (TDT) but
in patients with non -transfusion dependent thalassaemia (NTDT)
intermedia, idiopathic haemochromatosis, myelodyplasia, sickle cell
disease, post-transplanted sickle cell disease and thalassaemia as well
as many other categories of patients.[78,104-110]
With
regards to personalised medicine, the characterisation of the iron
metabolic or toxicity or other related targets is necessary for
designing the appropriate therapeutic strategies in each condition and
each patient, which can result in the optimisation of chelating drug
protocol or other therapeutic interventions.[111-117]
In this context, the mechanisms of iron release from ferritin and
haemosiderin, as well as other molecular or cellular mechanisms are of
particular interest.[118,119]
Changes in the
therapeutic strategies are necessary under special circumstances such
as pregnancy, splenomegaly, and infections and also when considering
the possible introduction of erythropoietic biological or other
emerging therapies.[120-123] Similar considerations
are in progress regarding other clinical issues such as the early
initiation of chelation therapy using L1 in thalassaemia children from
about one year of age and also the initiation of combination therapies.[124-127]
There are different criteria and opinions regarding the latter, with
the ICOC L1/DF combination, for example, to be available, safe and
flexible in all the patient categories and cases depending on the iron
load levels and the rate of body iron intake from transfusions, whereas
for other groups of investigators different restrictions are imposed in
the use of combination protocols (Figure 6).[9,57,58,73,81-83,99,100,103,128]
The
academic debates on the efficacy, toxicity, historical, and other
aspects of L1, DF and DFRA and their combinations are likely to
continue in the forthcoming years. Such debates are mostly focused on
past practises of ineffective therapies and not issues associated with
the current “golden era” period of iron chelation therapy in
thalassaemia, namely the achievement and maintenance of normal iron
stores.[81-83,128-131]
The
molecular, therapeutic, and other properties of L1 as a potent chelator
and antioxidant with access to most tissues and organs make it a unique
pharmaceutical with broad spectrum clinical applications.[75,95]
This prospect/dilemma is similar to that of the introduction of L1 as
the first oral iron chelating drug about 30 years ago and needs further
investigations to be confirmed.[132] Within this
context, specific therapeutic strategies have to be designed based on a
risk/benefit assessment for each condition and each patient. The aim
and targets of such therapeutic strategies need to be defined and
evaluated in a manner similar to the case of the paradigm of the
complete treatment of iron overload in thalassaemia using L1 and
selected L1/DF combinations.[58,81-83]
References
- Community control of hereditary anaemias: memorandum from a WHO meeting. Bull World Health Organ. 1983; 61: 63-80.
- Weatherall
DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global
health problem. Bull World Health Organ. 2001; 79: 704-12.
- Verma IC. Burden of genetic disorders in India. Indian J Pediatr. 2000; 67: 893-8. https://doi.org/10.1007/BF02723953 PMid:11262988
- Kontoghiorghes
GJ, Eracleous E, Economides C, Kolnagou A. Advances in iron overload
therapies. Prospects for effective use of deferiprone (L1),
deferoxamine, the new experimental chelators ICL670, GT56-252, L1NA11
and their combinations. Curr Med Chem. 2005; 12:2663-81. https://doi.org/10.2174/092986705774463003 PMid:16305464
- Telfer
PT, Warburton F, Christou S, Hadjigavriel M, Sitarou M, Kolnagou A,
Angastiniotis M. Improved survival in thalassemia major patients on
switching from desferrioxamine to combined chelation therapy with
desferrioxamine and deferiprone. Haematologica. 2009; 94: 1777-8. https://doi.org/10.3324/haematol.2009.009118 PMid:19815834 PMCid:PMC2791948
- Au
WY, Lee V, Lau CW, Yau J, Chan D, Chan EY, Cheung WW, Ha SY, Kho B, Lee
CY, Li RC, Li CK, Lin SY, Ling AS, Mak V, Sun L, Wong KH, Wong R, Yuen
HL. A synopsis of current care of thalassaemia major patients in Hong
Kong. Hong Kong Med J. 2011; 17: 261-6.
- Maggio
A, Filosa A, Vitrano A, Aloj G, Kattamis A, Ceci A, Fucharoen S,
Cianciulli P, Grady RW, Prossomariti L, Porter JB, Iacono A, Cappellini
MD, Bonifazi F, Cassarà F, Harmatz P, Wood J, Gluud C. Iron chelation
therapy in thalassemia major: a systematic review with meta-analyses of
1520 patients included on randomized clinical trials. Blood Cells Mol
Dis 2011; 47:166-75. https://doi.org/10.1016/j.bcmd.2011.07.002 PMid:21843958
- Kontoghiorghes GJ. The aim of iron chelation therapy in thalassaemia. Eur J Haematol. 2017;99:465-66. https://doi.org/10.1111/ejh.12939 PMid:28833560
- Tanner
MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, Pibiri M, Nair
SV, Walker JM, Pennell DJ. Combined chelation therapy in thalassemia
major for the treatment of severe myocardial siderosis with left
ventricular dysfunction. J Cardiovasc Magn Reson. 2008; 10: 12. https://doi.org/10.1186/1532-429X-10-12 PMid:18298856 PMCid:PMC2289829
- Maggio
A, Vitrano A, Lucania G, Capra M, Cuccia L, Gagliardotto F, Pitrolo L,
Prossomariti L, Filosa A, Caruso V, Gerardi C, Campisi S, Cianciulli P,
Rizzo M, D'Ascola G, Ciancio A, Di Maggio R, Calvaruso G, Pantalone GR,
Rigano P. Long-term use of deferiprone significantly enhances
left-ventricular ejection function in thalassemia major patients. Am J
Hematol. 2012;87:732-3. https://doi.org/10.1002/ajh.23219 PMid:22622672
- Boddaert
N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi
D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in
Friedreich ataxia: biologic and clinical implications. Blood. 2007;
110: 401-8. https://doi.org/10.1182/blood-2006-12-065433 PMid:17379741
- Abbruzzese
G, Cossu G, Balocco M, Marchese R, Murgia D, Melis M, Galanello R,
Barella S, Matta G, Ruffinengo U, Bonuccelli U, Forni GL. A pilot trial
of deferiprone for neurodegeneration with brain iron accumulation.
Haematologica. 2011; 96: 1708-11. https://doi.org/10.3324/haematol.2011.043018 PMid:21791473 PMCid:PMC3208690
- Cossu
G, Abbruzzese G, Matta G, Murgia D, Melis M, Ricchi V, Galanello R,
Barella S, Origa R, Balocco M, Pelosin E, Marchese R, Ruffinengo U,
Forni GL. Efficacy and safety of deferiprone for the treatment of
pantothenate kinase-associated neurodegeneration (PKAN) and
neurodegeneration with brain iron accumulation (NBIA): results from a
four years follow-up. Parkinsonism Relat Disord. 2014; 20: 651-4. https://doi.org/10.1016/j.parkreldis.2014.03.002 PMid:24661465
- Kontoghiorghes
GJ, Wilson MT. Structural and kinetic studies on eisenia foetida
erythrocruorin. In: J. Lamy ed. Invertebrate Oxygen Binding Proteins,
Structure, Active Site and Function. Marcell Dekker, New York and
Basel.1981, 385-391.
- Kontoghiorghes GJ
The design of orally active iron chelators for the treatment of
thalassaemia. PhD thesis, Colchester UK, University of Essex, British
Library Microfilm No D66194/86. 1982, pp 1-243. (https://www.pri.ac.cy/files/KGJ_thesis_1982.pdf)
- Kontoghiorghes
GJ. Design, properties and effective use of the oral chelator L1 and
other α-ketohydroxypyridines in the treatment of transfusional iron
overload in thalassaemia. Ann N Y Acad Sci. 1990; 612: 339-50. https://doi.org/10.1111/j.1749-6632.1990.tb24321.x PMid:2291562
- Kontoghiorghes GJ, Keast CG. A simple metabolic cage for mice. Laboratory practice. 1984;33: 90-1.
- Kontoghiorghes GJ, Marcus RE, Huehns ER. Desferrioxamine suppositories. Lancet. 1983; ii: 454. https://doi.org/10.1016/S0140-6736(83)90413-0
- Kontoghiorghes GJ. New orally active iron chelators. Lancet. 1985; i: 817. https://doi.org/10.1016/S0140-6736(85)91472-2
- Kontoghiorghes
GJ, Barr J, Nortey P, Sheppard L. Selection of a new generation of
orally active alpha-ketohydroxypyridine iron chelators intended for use
in the treatment of iron overload. Am J Hematol. 1993;42:340-9. https://doi.org/10.1002/ajh.2830420403 PMid:8493983
- Kontoghiorghes
GJ (ed). Oral chelation in the treatment of thalassaemia and other
diseases. Drugs Today. 1992; 28 (Suppl A):1-187.
- Kontoghiorghes
GJ, Pattichis K, Neocleous K, Kolnagou A. The design and development of
deferiprone (L1) and other iron chelators for clinical use: targeting
methods and application prospects. Curr Med Chem. 2004; 11: 2161-83. https://doi.org/10.2174/0929867043364685 PMid:15279556
- Kontoghiorghes
GJ, Sheppard L. Simple synthesis of the potent iron chelators
1-alkyl-3-hydroxy-2-methylpyrid-4-ones. Inorg Chim Acta. 1987;136:
L11-L12. https://doi.org/10.1016/S0020-1693(00)85549-8
- Pepe
A, Rossi G, Bentley A, Putti MC, Frizziero L, D'Ascola DG, Cuccia L,
Spasiano A, Filosa A, Caruso V, Hanif A, Meloni A. Cost-Utility
Analysis of Three Iron Chelators Used in Monotherapy for the Treatment
of Chronic Iron Overload in β-Thalassaemia Major Patients: An Italian
Perspective. Clin Drug Investig. 2017; 37:453-64. https://doi.org/10.1007/s40261-017-0496-1 PMid:28185140
- Li
J., Lin Y., Li X., Zhang J. Economic evaluation of chelation regimens
for β-thalassemia major: a systematic review. Mediterr J Hematol Infect
Dis. 2019, 11: e2019036 https://doi.org/10.4084/mjhid.2019.036 PMid:31308912 PMCid:PMC6613630
- Luangasanatip
N, Chaiyakunapruk N, Upakdee N, Wong P. Iron-chelating therapies in a
transfusion-dependent thalassaemia population in Thailand: a
cost-effectiveness study. Clin Drug Investig. 2011;31:493-505. https://doi.org/10.2165/11587120-000000000-00000 PMid:21627338
- Viprakasit
V, Nuchprayoon I, Chuansumrit A, Torcharus K, Pongtanakul B,
Laothamatas J, Srichairatanakool S, Pooliam J, Supajitkasem S,
Suriyaphol P, Tanphaichitr VS, Tuchinda S. Deferiprone (GPO-L-ONE(®))
monotherapy reduces iron overload in transfusion-dependent
thalassemias: 1-year results from a multicenter prospective, single
arm, open label, dose escalating phase III pediatric study (GPO-L-ONE;
A001) from Thailand. Am J Hematol. 2013;88:251-60. https://doi.org/10.1002/ajh.23386 PMid:23460233
- Kontoghiorghes G, Hider RC, Silver J. Pharmaceutical compositions. British patent specification number 8208608.1982.
- Kontoghiorghes
GJ, Hoffbrand AV, Hider RC, Huehns ER, Charalambous J. New orally
active iron chelators. Br J Haematol. 1985; 64: A61, p567.
- Hider RC, Kontoghiorghes G, Silver J. Pharmaceutical compositions: UK Patent GB2118176, 1983.
- Gyparaki
M, Porter JB, Hirani S, Streater M, Hider RC, Huehns ER. In vivo
evaluation of hydroxypyridone iron chelators in a mouse model. Acta
Haematol. 1987;78:217-21. https://doi.org/10.1159/000205878 PMid:3120475
- Huehns
ER, Porter JB, Hider RC. Selection of hydroxypyridin-4-ones for the
treatment of iron overload using in vitro and in vivo models.
Hemoglobin. 1988;12:593-600. https://doi.org/10.3109/03630268808991649 PMid:3209401
- Porter
JB, Gyparaki M, Burke LC, Huehns ER, Sarpong P, Saez V, Hider RC. Iron
mobilization from hepatocyte monolayer cultures by chelators: the
importance of membrane permeability and the iron-binding constant.
Blood. 1988;72:1497-503. https://doi.org/10.1182/blood.V72.5.1497.1497 PMid:3179437
- Hider
RC, Singh S, Porter JB, Huehns ER. The development of
hydroxypyridin-4-ones as orally active iron chelators. Ann N Y Acad
Sci. 1990;612:327-38. https://doi.org/10.1111/j.1749-6632.1990.tb24320.x PMid:2291561
- Porter
JB, Morgan J, Hoyes KP, Burke LC, Huehns ER, Hider RC. Relative oral
efficacy and acute toxicity of hydroxypyridin-4-one iron chelators in
mice. Blood. 1990;76:2389-96. https://doi.org/10.1182/blood.V76.11.2389.2389 PMid:2257308
- Porter JB, Hoyes KP, Abeysinghe R, Huehns ER, Hider RC. Animal toxicology of iron chelator L1. Lancet. 1989;2(8655):156. https://doi.org/10.1016/S0140-6736(89)90206-7
- Porter
JB, Abeysinghe RD, Hoyes KP, Barra C, Huehns ER, Brooks PN, Blackwell
MP, Araneta M, Brittenham G, Singh S, Dobbin P, Hider RC. Contrasting
interspecies efficacy and toxicology of
1,2-diethyl-3-hydroxypyridin-4-one, CP94, relates to differing
metabolism of the iron chelating site. Br J Haematol. 1993;85:159-68. https://doi.org/10.1111/j.1365-2141.1993.tb08660.x PMid:8251385
- Epemolu
RO, Ackerman R, Porter JB, Hider RC, Damani LA, Singh S. HPLC
determination of 1,2-diethyl-3-hydroxypyridin-4-one (CP94), its iron
complex [Fe(III) (CP94)3] and glucuronide conjugate [CP94-GLUC] in
serum and urine of thalassaemic patients. J Pharm Biomed Anal.
1994;12:923-30. https://doi.org/10.1016/0731-7085(94)E0027-X
- Porter
JB, Singh S, Hoyes KP, Epemolu O, Abeysinghe RD, Hider RC. Lessons from
preclinical and clinical studies with
1,2-diethyl-3-hydroxypyridin-4-one, CP94 and related compounds. Adv Exp
Med Biol. 1994;356:361-70. https://doi.org/10.1007/978-1-4615-2554-7_38 PMid:7887242
- Pfannkuch F, Bentley P, Schnebli HP. Future of oral iron chelator deferiprone (L1) Lancet. 1993;341(8858):1480. https://doi.org/10.1016/0140-6736(93)90925-7
- Hershko C. Development of oral iron chelator L1. Lancet. 1993;341(8852):1088-9. https://doi.org/10.1016/0140-6736(93)92444-X
- Savulescu J. Thalassaemia major: the murky story of deferiprone. Br Med J 2004;328(7436):358-9. https://doi.org/10.1136/bmj.328.7436.358 PMid:14962851 PMCid:PMC341373
- REUTERS [webpage on the Internet]. U.S. seeks up to $3.35 billion in Novartis kickback lawsuit; 2015. Available from: http://www.reuters.com/article/2015/06/30/us-novartis-lawsuit-idUSKCN0-PA1ZK20150630. Accessed October 1, 2019.
- Kontoghiorghe
CN, Kontoghiorghes GJ. New developments and controversies in iron
metabolism and iron chelation therapy. World J Methodol. 2016;6:1-19. https://doi.org/10.5662/wjm.v6.i1.1 PMid:27019793 PMCid:PMC4804243
- Kontoghiorghe
CN, Andreou N, Constantinou K, Kontoghiorghes GJ. World health
dilemmas: orphan and rare diseases, orphan drugs and orphan patients.
World J Methodol. 2014;4:163-88. https://doi.org/10.5662/wjm.v4.i3.163 PMid:25332915 PMCid:PMC4202455
- Kontoghiorghes
GJ, Aldouri MA, Sheppard L, Hoffbrand AV.
1,2-Dimethyl-3-hydroxypyrid-4-one, an orally active chelator for
treatment of iron overload. Lancet. 1987; 1: 1294-5. https://doi.org/10.1016/S0140-6736(87)90545-9
- Kontoghiorghes
GJ, Aldouri MA, Hoffbrand AV, Barr J, Wonke B, Kourouclaris T, Sheppard
L. Effective chelation of iron in beta thalassaemia with the oral
chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Br Med J (Clin Res Ed).
1987; 295: 1509-12. https://doi.org/10.1136/bmj.295.6612.1509 PMid:3122880 PMCid:PMC1248663
- Hoffbrand
AV, Bartlett AN, Veys PA, O'Connor NT, Kontoghiorghes GJ.
Agranulocytosis and thrombocytopenia in patient with Blackfan-Diamond
anaemia during oral chelator trial. Lancet. 1989; 2(8660):457. https://doi.org/10.1016/S0140-6736(89)90641-7
- The history of deferiprone. https://www.youtube.com/watch?v=ZcvSLyIgYd8. Accessed October 1, 2019.
- Agarwal
MB, Gupte SS, Viswanathan C, Vasandani D, Ramanathan J, Desai N,
Puniyani RR, Chhablani AT. Long-term assessment of efficacy and safety
of L1, an oral iron chelator, in transfusion dependent thalassaemia:
Indian trial. Br J Haematol. 1992;82:460-6. https://doi.org/10.1111/j.1365-2141.1992.tb06445.x PMid:1419829
- Cohen
AR, Galanello R, Piga A, De Sanctis V, Tricta F. Safety and
effectiveness of long-term therapy with the oral iron chelator
deferiprone. Blood. 2003;102:1583-7. https://doi.org/10.1182/blood-2002-10-3280 PMid:12763939
- Kontoghiorghes
GJ, Goddard JG, Bartlett AN, Sheppard L. Pharmacokinetic studies in
humans with the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one.
Clin Pharmacol Ther. 1990; 48: 255-61. https://doi.org/10.1038/clpt.1990.147 PMid:2401124
- Kontoghiorghes
GJ, Bartlett AN, Hoffbrand AV, Goddard JG, Sheppard L, Barr J, Nortey
P. Long-term trial with the oral iron chelator
1,2-dimethyl-3-hydroxypyrid-4-one (L1). I. Iron chelation and metabolic
studies. Br J Haematol. 1990; 76: 295-300. https://doi.org/10.1111/j.1365-2141.1990.tb07887.x PMid:2094333
- Fassos
FF, Klein J, Fernandes D, Matsui D, Olivieri NF, Koren G. The
pharmacokinetics and pharmacodynamics of the oral iron chelator
deferiprone (L1) in relation to hemoglobin levels. Int J Clin Pharmacol
Ther. 1996;34:288-92.
- Nielsen P, Frtjes
M, Drescow B, Fischer R, Engelhardt R, Heinrich H C. The iron
decorporation effect of L1 in normal and TMH-Ferrocene iron-loaded rats
and in one patient with post-transfusional siderosis as judged by Fe 59
-labelling technique. Drugs Today. 1992; 28 (Suppl A): 45-53.
- Kontoghiorghes
GJ. Iron mobilization from transferrin and non-transferrin-bound-iron
by deferiprone. Implications in the treatment of thalassaemia, anemia
of chronic disease, cancer and other conditions. Hemoglobin.
2006;30:183-200. https://doi.org/10.1080/03630260600642450 PMid:16798643
- Di
Maggio R, Maggio A.The new era of chelation treatments: effectiveness
and safety of 10 different regimens for controlling iron overloading in
thalassaemia major. Br J Haematol. 2017;178:676-88. https://doi.org/10.1111/bjh.14712 PMid:28439891
- Kolnagou
A, Kontoghiorghes GJ. Chelation protocols for the elimination and
prevention of iron overload in thalassaemia. Front Biosci (Landmark
Ed). 2018;23:1082-98. https://doi.org/10.2741/4634 PMid:28930590
- Songdej
D, Sirachainan N, Wongwerawattanakoon P, Sasanakul W, Kadegasem P,
Sungkarat W, Chuansumrit A. Combined chelation therapy with daily oral
deferiprone and twice-weekly subcutaneous infusion of desferrioxamine
in children with β-thalassemia: 3-year experience. Acta Haematol.
2015;133:226-36. https://doi.org/10.1159/000363210 PMid:25376266
- Pepe
A, Meloni A, Pistoia L, Cuccia L, Gamberini MR, Lisi R, D'Ascola DG,
Rosso R, Allò M, Spasiano A, Restaino G, Righi R, Mangione M, Positano
V, Ricchi P. MRI multicentre prospective survey in thalassaemia major
patients treated with deferasirox versus deferiprone and
desferrioxamine. Br J Haematol. 2018;183:783-95. https://doi.org/10.1111/bjh.15595 PMid:30334574
- Kontoghiorghes
GJ. A new era in iron chelation therapy: the design of optimal,
individually adjusted iron chelation therapies for the complete removal
of iron overload in thalassemia and other chronically transfused
patients. Hemoglobin. 2009; 33:332-8. https://doi.org/10.3109/03630260903217182 PMid:19814679
- Filosa
A, Vitrano A, Rigano P, Calvaruso G, Barone R, Capra M, Cuccia L,
Gagliardotto F, Pitrolo L, Prossomariti L, Casale M, Caruso V, Gerardi
C, Campisi S, Cianciulli P, Rizzo M, D'Ascola G, Ciancio A, Maggio A.
Long-term treatment with deferiprone enhances left ventricular ejection
function when compared to deferoxamine in patients with thalassemia
major. Blood Cells Mol Dis. 2013;51:85-8. https://doi.org/10.1016/j.bcmd.2013.04.002 PMid:23628348
- Smeets
ME, Vreugdenhil G, Holdrinet RS. Improvement of erythropoiesis during
treatment with deferiprone in a patient with myelofibrosis and
transfusional hemosiderosis. Am J Hematol. 1996 ;51:243-4. https://doi.org/10.1002/(SICI)1096-8652(199603)51:3<243::AID-AJH12>3.0.CO;2-H
- Chang
YH, Shaw CF, Wu KH, Hsieh KH, Su YN, Lu PJ. Treatment with deferiprone
for iron overload alleviates bone marrow failure in a Fanconi anemia
patient. Hemoglobin. 2009;33:346-51. https://doi.org/10.3109/03630260903212563 PMid:19814681
- Sriwantana
T, Vivithanaporn P, Paiboonsukwong K, Rattanawonsakul K, Srihirun S,
Sibmooh N. Deferiprone increases endothelial nitric oxide synthase
phosphorylation and nitric oxide production. Can J Physiol Pharmacol.
2018;96:879-85. https://doi.org/10.1139/cjpp-2018-0012 PMid:29806986
- Zurlo
MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C,
Di Gregorio F, Burattini MG, Terzoli S. Survival and causes of death in
thalassaemia major. Lancet. 1989; 2: 27-30. https://doi.org/10.1016/S0140-6736(89)90264-X
- Kyriacou
K, Michaelides Y, Senkus R, Simamonian K, Pavlides N, Antoniades L,
Zambartas C. Ultrastructural pathology of the heart in patients with
beta-thalassaemia major. Ultrastruct Pathol. 2000; 24: 75-81. https://doi.org/10.1080/01913120050118549 PMid:10808552
- Kolnagou
A, Economides C, Eracleous E, Kontoghiorghes GJ. Low serum ferritin
levels are misleading for detecting cardiac iron overload and increase
the risk of cardiomyopathy in thalassemia patients. The importance of
cardiac iron overload monitoring using magnetic resonance imaging T2
and T2*. Hemoglobin. 2006;30:219-27. https://doi.org/10.1080/03630260600642542 PMid:16798647
- Papakonstantinou
O, Alexopoulou E, Economopoulos N, Benekos O, Kattamis A, Kostaridou S,
Ladis V, Efstathopoulos E, Gouliamos A, Kelekis NL. Assessment of iron
distribution between liver, spleen, pancreas, bone marrow, and
myocardium by means of R2 relaxometry with MRI in patients with
beta-thalassemia major. J Magn Reson Imaging. 2009;29:853-9. https://doi.org/10.1002/jmri.21707 PMid:19306409
- Kolnagou
A, Natsiopoulos K, Kleanthous M, Ioannou A, Kontoghiorghes GJ. Liver
iron and serum ferritin levels are misleading for estimating cardiac,
pancreatic, splenic and total body iron load in thalassemia patients:
factors influencing the heterogenic distribution of excess storage iron
in organs as identified by MRI T2*. Toxicol Mech Methods. 2013; 23:
48-56. https://doi.org/10.3109/15376516.2012.727198 PMid:22943064
- Mavrogeni
SI, Gotsis ED, Markussis V, Tsekos N, Politis C, Vretou E, Kermastinos
D. T2 relaxation time study of iron overload in b-thalassemia. MAGMA.
1998;6:7-12. https://doi.org/10.1007/BF02662506 PMid:9794284
- Anderson
LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, Firmin DN,
Wonke B, Porter J, Walker JM, Pennell DJ. Cardiovascular T2-star (T2*)
magnetic resonance for the early diagnosis of myocardial iron overload.
Eur Heart J. 2001;22: 2171-9. https://doi.org/10.1053/euhj.2001.2822 PMid:11913479
- Kolnagou
A, Yazman D, Economides C, Eracleous E, Kontoghiorghes GJ. Uses and
limitations of serum ferritin, magnetic resonance imaging T2 and T2* in
the diagnosis of iron overload and in the ferrikinetics of
normalization of the iron stores in thalassemia using the International
Committee on Chelation deferiprone/deferoxamine combination protocol.
Hemoglobin. 2009;33:312-22. https://doi.org/10.3109/03630260903213231 PMid:19814677
- Kontoghiorghes
GJ, Neocleous K, Kolnagou A. Benefits and risks of deferiprone in iron
overload in thalassaemia and other conditions: comparison of
epidemiological and therapeutic aspects with deferoxamine. Drug Safety.
2003;26:553-84. https://doi.org/10.2165/00002018-200326080-00003 PMid:12825969
- Kontoghiorghes
GJ, Kontoghiorghe CN. Prospects for the introduction of targeted
antioxidant drugs for the prevention and treatment of diseases related
to free radical pathology. Expert Opin Investig Drugs. 2019;28:
593-603. https://doi.org/10.1080/13543784.2019.1631284 PMid:31185180
- Modell
B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK:
data from the UK Thalassaemia Register. Lancet. 2000; 355: 2051-2. https://doi.org/10.1016/S0140-6736(00)02357-6
- Kolnagou
A, Kontoghiorghe CN, Kontoghiorghes GJ. Transition of thalassaemia and
Friedreich ataxia from fatal to chronic diseases. World J Methodol.
2014;4:197-218. https://doi.org/10.5662/wjm.v4.i4.197 PMid:25541601 PMCid:PMC4274580
- Kontoghiorghe
CN, Kontoghiorghes GJ. Efficacy and safety of iron-chelation therapy
with deferoxamine, deferiprone, and deferasirox for the treatment of
iron-loaded patients with non-transfusion-dependent thalassemia
syndromes. Drug Des Devel Ther. 2016;10: 465-81. https://doi.org/10.2147/DDDT.S79458 PMid:26893541 PMCid:PMC4745840
- Kontoghiorghes GJ. Advances in oral iron chelation in man. Int J Hematol. 1992;55: 27-38.
- Kolnagou
A, Michaelides Y, Kontos C, Kyriacou K, Kontoghiorghes GJ. Myocyte
damage and loss of myofibers is the potential mechanism of iron
overload toxicity in congestive cardiac failure in thalassemia.
Complete reversal of the cardiomyopathy and normalization of iron load
by deferiprone. Hemoglobin. 2008; 32: 17-28. https://doi.org/10.1080/03630260701726491 PMid:18274979
- Kolnagou
A, Kleanthous M, Kontoghiorghes GJ. Reduction of body iron stores to
normal range levels in thalassaemia by using a deferiprone/deferoxamine
combination and their maintenance thereafter by deferiprone
monotherapy. Eur J Haematol. 2010; 85: 430-8. https://doi.org/10.1111/j.1600-0609.2010.01499.x PMid:20662901
- Haematol. 2010; 148: 466-75. https://doi.org/10.1111/j.1365-2141.2009.07970.x PMid:19912219
- Kolnagou
A, Kontoghiorghe CN, Kontoghiorghes GJ. Prevention of Iron Overload and
Long Term Maintenance of Normal Iron Stores in Thalassaemia Major
Patients using Deferiprone or Deferiprone Deferoxamine Combination.
Drug Res (Stuttg). 2017; 67:404-11. https://doi.org/10.1055/s-0043-102691 PMid:28320041
- Aessopos
A, Kati M, Farmakis D, Polonifi E, Deftereos S, Tsironi M. Intensive
chelation therapy in beta-thalassemia and possible adverse cardiac
effects of desferrioxamine. Int J Hematol. 2007;86:212-5. https://doi.org/10.1007/BF03006922 PMid:17988985
- Konstantinou
E, Pashalidis I, Kolnagou A, Kontoghiorghes GJ. Interactions of
hydroxycarbamide (hydroxyurea) with iron and copper: implications on
toxicity and therapeutic strategies. Hemoglobin. 2011;35:237-46. https://doi.org/10.3109/03630269.2011.578950 PMid:21599436
- Rajapurkar
MM, Hegde U, Bhattacharya A, Alam MG, Shah SV. Effect of deferiprone,
an oral iron chelator, in diabetic and non-diabetic glomerular disease.
Toxicol Mech Methods. 2013; 23: 5-10. https://doi.org/10.3109/15376516.2012.730558 PMid:22978744
- Mohanty
D, Ghosh K, Pathare AV, Karnad D. Deferiprone (L1) as an adjuvant
therapy for Plasmodium falciparum malaria. Indian J Med Res. 2002; 115:
17-21.
- Martin-Bastida A, Ward RJ,
Newbould R, Piccini P, Sharp D, Kabba C, Patel MC, Spino M, Connelly J,
Tricta F, Crichton RR, Dexter DT. Brain iron chelation by deferiprone
in a phase 2 randomised double-blinded placebo controlled clinical
trial in Parkinson's disease. Sci Rep. 2017;7:1398. https://doi.org/10.1038/s41598-017-01402-2 PMid:28469157 PMCid:PMC5431100
- Saxena
D, Spino M, Tricta F, Connelly J, Cracchiolo BM, Hanauske AR.
Drug-Based Lead Discovery: The Novel Ablative Antiretroviral Profile of
Deferiprone in HIV-1-Infected Cells and in HIVInfected Treatment-Naive
Subjects of a Double-Blind, Placebo-Controlled, Randomized Exploratory
Trial. PLoS One. 2016;11:e0154842. https://doi.org/10.1371/journal.pone.0154842 PMid:27191165 PMCid:PMC4871512
- Leftin
A, Zhao H, Turkekul M, de Stanchina E, Manova K, Koutcher JA. Iron
deposition is associated with differential macrophage infiltration and
therapeutic response to iron chelation in prostate cancer. Sci Rep.
2017;7:11632. https://doi.org/10.1038/s41598-017-11899-2 PMid:28912459 PMCid:PMC5599545
- Weigel KJ, Lynch SG, Levine SM. Iron chelation and multiple sclerosis. ASN Neuro. 2014; 6: e00136. https://doi.org/10.1042/AN20130037 PMid:24397846 PMCid:PMC3906635
- Barnabé
N, Zastre JA, Venkataram S, Hasinoff BB. Deferiprone protects against
doxorubicin-induced myocyte cytotoxicity. Free Radic Biol Med. 2002;33:
266-75. https://doi.org/10.1016/S0891-5849(02)00873-0
- Ueda
K, Kim HJ, Zhao J, Song Y, Dunaief JL, Sparrow JR. Iron promotes
oxidative cell death caused by bisretinoids of retina. Proc Natl Acad
Sci U S A. 2018;115: 4963-8. https://doi.org/10.1073/pnas.1722601115 PMid:29686088 PMCid:PMC5948992
- Maher
P, Kontoghiorghes GJ. Characterization of the neuroprotective potential
of derivatives of the iron chelating drug deferiprone. Neurochem Res
2015;40: 609-20. https://doi.org/10.1007/s11064-014-1508-7 PMid:25559767
- Kontoghiorghe
CN, Kolnagou A, Kontoghiorghes GJ. Antioxidant targeting by deferiprone
in diseases related to oxidative damage. Front Biosci (Landmark Ed).
2014;19: 862-85. https://doi.org/10.2741/4253 PMid:24896322
- Origa
R, Danjou F, Cossa S, Matta G, Bina P, Dessì C, Defraia E, Foschini ML,
Leoni G, Morittu M, Galanello R. Impact of heart magnetic resonance
imaging on chelation choices, compliance with treatment and risk of
heart disease in patients with thalassaemia major. Br J Haematol.
2013;163:400-3. https://doi.org/10.1111/bjh.12517 PMid:24033185
- Platis
O, Anagnostopoulos G, Farmaki K, Posantzis M, Gotsis E, Tolis G.
Glucose metabolism disorders improvement in patients with thalassaemia
major after 24-36 months of intensive chelation therapy. Pediatr
Endocrinol Rev. 2004; 2 Suppl 2: 279-81.
- Borgna-Pignatti
C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR,
Ghilardi R, Piga A, Romeo MA, Zhao H, Cnaan A. Cardiac morbidity and
mortality in deferoxamine- or deferiprone-treated patients with
thalassemia major. Blood. 2006; 107: 3733-7. https://doi.org/10.1182/blood-2005-07-2933 PMid:16373663
- Tanner
MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, Pibiri M, Nair
SV, Walker JM, Pennell DJ. Combined chelation therapy in thalassemia
major for the treatment of severe myocardial siderosis with left
ventricular dysfunction. J Cardiovasc Magn Reson. 2008; 10: 12. https://doi.org/10.1186/1532-429X-10-12 PMid:18298856 PMCid:PMC2289829
- Pepe
A, Meloni A, Rossi G, Cuccia L, D'Ascola GD, Santodirocco M, Cianciulli
P, Caruso V, Romeo MA, Filosa A, Pitrolo L, Putti MC, Peluso A, Campisi
S, Missere M, Midiri M, Gulino L, Positano V, Lombardi M, Ricchi P.
Cardiac and hepatic iron and ejection fraction in thalassemia major:
multicentre prospective comparison of combined deferiprone and
deferoxamine therapy against deferiprone or deferoxamine monotherapy. J
Cardiovasc Magn Reson. 2013;15:1. https://doi.org/10.1186/1532-429X-15-1 PMid:23324167 PMCid:PMC3599638
- Pepe
A, Meloni A, Rossi G, Midiri M, Missere M, Valeri G, Sorrentino F,
D'Ascola DG, Spasiano A, Filosa A, Cuccia L, Dello Iacono N, Forni G,
Caruso V, Maggio A, Pitrolo L, Peluso A, De Marchi D, Positano V, Wood
JC. Prediction of cardiac complications for thalassemia major in the
widespread cardiac magnetic resonance era: a prospective multicentre
study by a multi-parametric approach. Eur Heart J Cardiovasc Imaging.
2018;19:299-309. https://doi.org/10.1093/ehjci/jex012 PMid:28200076
- Pepe
A, Meloni A, Capra M, Cianciulli P, Prossomariti L, Malaventura C,
Putti MC, Lippi A, Romeo MA, Bisconte MG, Filosa A, Caruso V, Quarta A,
Pitrolo L, Missere M, Midiri M, Rossi G, Positano V, Lombardi M, Maggio
A. Deferasirox, deferiprone and desferrioxamine treatment in
thalassemia major patients: cardiac iron and function comparison
determined by quantitative magnetic resonance imaging. Haematologica.
2011;96:41-7. https://doi.org/10.3324/haematol.2009.019042 PMid:20884710 PMCid:PMC3012763
- Pennell
DJ, Udelson JE, Arai AE, Bozkurt B, Cohen AR, Galanello R, Hoffman TM,
Kiernan MS, Lerakis S, Piga A, Porter JB, Walker JM, Wood J;
Cardiovascular function and treatment in β-thalassemia major: a
consensus statement from the American Heart Association. Circulation.
2013;128: 281-308. https://doi.org/10.1161/CIR.0b013e31829b2be6 PMid:23775258
- Steensma DP. Myelodysplasia paranoia: iron as the new radon. Leuk Res. 2009; 33:1158-63. https://doi.org/10.1016/j.leukres.2008.10.017 PMid:19036443
- Lucania
G, Vitrano A, Filosa A, Maggio A. Chelation treatment in
sickle-cell-anaemia: much ado about nothing? Br J Haematol. 2011; 154:
545-55. https://doi.org/10.1111/j.1365-2141.2011.08769.x PMid:21707578
- Marsella
M, Borgna-Pignatti C.Transfusional iron overload and iron chelation
therapy in thalassemia major and sickle cell disease. Hematol Oncol
Clin North Am. 2014;28:703-27. https://doi.org/10.1016/j.hoc.2014.04.004 PMid:25064709
- Lucarelli
G, Angelucci E, Giardini C, Baronciani D, Galimberti M, Polchi P,
Bartolucci M, Muretto P, Albertini F. Fate of iron stores in
thalassaemia after bone-marrow transplantation. Lancet.
1993;342(8884):1388-91. https://doi.org/10.1016/0140-6736(93)92753-G
- Pilo
F, Angelucci E. Iron Toxicity and Hemopoietic Cell Transplantation:
Time to Change the Paradigm. Mediterr J Hematol Infect Dis.
2019;11:e2019030. https://doi.org/10.4084/mjhid.2019.030 PMid:31205634 PMCid:PMC6548208
- Sharma D C. Patent rulings raise hope for cheap cancer drugs in India. Lancet Oncology. 2013; 14: e441. https://doi.org/10.1016/S1470-2045(13)70395-4
- Luangasanatip
N, Chaiyakunapruk N, Upakdee N, Wong P. Iron-chelating therapies in a
transfusion-dependent thalassaemia population in Thailand: a
cost-effectiveness study. Clin Drug Investig. 2011; 31:493-505. https://doi.org/10.2165/11587120-000000000-00000 PMid:21627338
- Szumowski
J, Bas E, Gaarder K, Schwarz E, Erdogmus D, Hayflick S. Measurement of
brain iron distribution in Hallevorden-Spatz syndrome. J Magn Reson
Imaging. 2010;31:482-9. https://doi.org/10.1002/jmri.22031 PMid:20099363
- Barbosa
JH, Santos AC, Tumas V, Liu M, Zheng W, Haacke EM, Salmon CE.
Quantifying brain iron deposition in patients with Parkinson's disease
using quantitative susceptibility mapping, R2 and R2*. Magn Reson
Imaging. 2015;33:559-65. https://doi.org/10.1016/j.mri.2015.02.021 PMid:25721997
- Dashtipour
K, Liu M, Kani C, Dalaie P, Obenaus A, Simmons D, Gatto NM, Zarifi
M.Iron Accumulation Is Not Homogenous among Patients with Parkinson's
Disease. Parkinsons Dis. 2015;2015:324843. https://doi.org/10.1155/2015/324843 PMid:25945281 PMCid:PMC4402185
- Kolnagou
A, Kleanthous M, Kontoghiorghes GJ. Efficacy, compliance and toxicity
factors are affecting the rate of normalization of body iron stores in
thalassemia patients using the deferiprone and deferoxamine combination
therapy. Hemoglobin. 2011;35:186-98. https://doi.org/10.3109/03630269.2011.576153 PMid:21599431
- Binding
A, Ward R, Tomlinson G, Kuo KHM. Deferiprone exerts a dose-dependent
reduction of liver iron in adults with iron overload. Eur J Haematol.
2019; 103: 80-7. https://doi.org/10.1111/ejh.13244 PMid:31066943
- Klopstock
T, Tricta F, Neumayr L, Karin I, Zorzi G, Fradette C, Kmieć T, Büchner
B, Steele HE, Horvath R, Chinnery PF, Basu A, Küpper C, Neuhofer C,
Kálmán B, Dušek P, Yapici Z, Wilson I, Zhao F, Zibordi F, Nardocci N,
Aguilar C, Hayflick SJ, Spino M, Blamire AM, Hogarth P, Vichinsky E.
Safety and efficacy of deferiprone for pantothenate kinase-associated
neurodegeneration: a randomised, double-blind, controlled trial and an
open-label extension study. Lancet Neurol. 2019;18: 631-42.https://doi.org/10.1016/S1474-4422(19)30142-5
- Kolnagou
A, Kontoghiorghe CN, Kontoghiorghes GJ. New targeted therapies and
diagnostic methods for iron overload diseases. Front Biosci (Schol Ed).
2018;10:1-20. https://doi.org/10.2741/s498 PMid:28930516
- La
A, Nguyen T, Tran K, Sauble E, Tu D, Gonzalez A, Kidane TZ, Soriano C,
Morgan J, Doan M, Tran K, Wang CY, Knutson MD, Linder MC. Mobilization
of iron from ferritin: new steps and details. Metallomics.
2018;10:154-68. https://doi.org/10.1039/C7MT00284J PMid:29260183
- Kontoghiorghes
GJ. Iron mobilization from ferritin using alpha-oxohydroxy
heteroaromatic chelators. Biochem J. 1986 ;233:299-302. https://doi.org/10.1042/bj2330299 PMid:3954731 PMCid:PMC1153022
- Origa R, Comitini F. Pregnancy in Thalassemia. Mediterr J Hematol Infect Dis. 2019;11:e2019019. https://doi.org/10.4084/mjhid.2019.019 PMid:30858957 PMCid:PMC6402552
- Kolnagou
A, Kontoghiorghe CN, Kontoghiorghes GJ. Transfusion-Related Acute Lung
Injury (TRALI) in two Thalassaemia Patients Caused by the Same
Multiparous Blood Donor. Mediterr J Hematol Infect Dis.
2017;9:e2017060. https://doi.org/10.4084/mjhid.2017.060 PMid:29181137 PMCid:PMC5667526
- Kolnagou
A, Michaelides Y, Kontoghiorghe CN, Kontoghiorghes GJ. The importance
of spleen, spleen iron, and splenectomy for determining total body iron
load, ferrikinetics, and iron toxicity in thalassemia major patients.
Toxicol Mech Methods. 2013;23:34-41. https://doi.org/10.3109/15376516.2012.735278 PMid:23039902
- Piga
A, Perrotta S, Gamberini MR, Voskaridou E, Melpignano A, Filosa A,
Caruso V, Pietrangelo A, Longo F, Tartaglione I, Borgna-Pignatti C,
Zhang X, Laadem A, Sherman ML, Attie KM. Luspatercept improves
hemoglobin levels and blood transfusion requirements in a study of
patients with β-thalassemia. Blood. 2019;133:1279-89. https://doi.org/10.1182/blood-2018-10-879247 PMid:30617198 PMCid:PMC6440118
- Elalfy
MS, Adly A, Awad H, Tarif Salam M, Berdoukas V, Tricta F. Safety and
efficacy of early start of iron chelation therapy with deferiprone in
young children newly diagnosed with transfusion-dependent thalassemia:
A randomized controlled trial. Am J Hematol. 2018;93:262-8. https://doi.org/10.1002/ajh.24966 PMid:29119631
- Chuansumrit
A, Songdej D, Sirachainan N, Wongwerawattanakoon P, Kadegasem P,
Sasanakul W. Safety profile of a liquid formulation of deferiprone in
young children with transfusion-induced iron overload: a 1-year
experience. Paediatr Int Child Health. 2016: 36:209-13. https://doi.org/10.1179/2046905515Y.0000000040 PMid:26052612
- Makis
A, Chaliasos N, Alfantaki S, Karagouni P, Siamopoulou A. Chelation
therapy with oral solution of deferiprone in transfusional
iron-overloaded children with hemoglobinopathies. Anemia.
2013;2013:121762. https://doi.org/10.1155/2013/121762 PMid:24294523 PMCid:PMC3835355
- Naithani
R, Chandra J, Sharma S. Safety of oral iron chelator deferiprone in
young thalassaemics. Eur J Haematol. 2005;74:217-20. https://doi.org/10.1111/j.1600-0609.2004.00377.x PMid:15693791
- Lin
CH, Chen X, Wu CC, Wu KH, Song TS, Weng TF, Hsieh YW, Peng CT.
Therapeutic mechanism of combined oral chelation therapy to maximize
efficacy of iron removal in transfusion-dependent thalassemia major - a
pilot study. Expert Rev Hematol. 2019;12:265-72. https://doi.org/10.1080/17474086.2019.1593823 PMid:30920854
- Olivieri
NF, Sabouhanian A, Gallie BL. Single-center retrospective study of the
effectiveness and toxicity of the oral iron chelating drugs deferiprone
and deferasirox. PLoS One. 2019;14:e0211942. https://doi.org/10.1371/journal.pone.0211942 PMid:30811439 PMCid:PMC6392256
- Hider RC, Hoffbrand AV. The Role of Deferiprone in Iron Chelation. N Engl J Med. 2018;379:2140-50. https://doi.org/10.1056/NEJMra1800219 PMid:30485781
- Kolnagou
A, Kontoghiorghes GJ. New golden era of chelation therapy in
thalassaemia: the achievement and maintenance of normal range body iron
stores. Br J Haematol. 2010;150:489-90. https://doi.org/10.1111/j.1365-2141.2010.08229.x PMid:20507309
- Kontoghiorghes GJ. Oral iron chelation is here. Br Med J. 991;303: 1279-80. https://doi.org/10.1136/bmj.303.6813.1279 PMid:1747667 PMCid:PMC1671411
- Galanello
R, Piga A, Alberti D, Rouan MC, Bigler H, Séchaud R. Safety,
tolerability, and pharmacokinetics of ICL670, a new orally active
iron-chelating agent in patients with transfusion-dependent iron
overload due to beta-thalassemia. J Clin Pharmacol. 2003;43:565-72. https://doi.org/10.1177/0091270003253350 PMid:12817519
- Kontoghiorghes GJ. Chelators affecting iron absorption in mice. Arzneimittelforschung. 1990;40:1332-5
- Kontoghiorghes
GJ, Spyrou A, Kolnagou A. Iron chelation therapy in hereditary
hemochromatosis and thalassemia intermedia: regulatory and non
regulatory mechanisms of increased iron absorption. Hemoglobin.
2010;34:251-64. https://doi.org/10.3109/03630269.2010.486335 PMid:20524815
- Schmidt
C, Ahmad T, Tulassay Z, Baumgart DC, Bokemeyer B, Howaldt S, Stallmach
A1, Büning C7; AEGIS Study Group. Ferric maltol therapy for iron
deficiency anaemia in patients with inflammatory bowel disease:
long-term extension data from a Phase 3 study. Aliment Pharmacol Ther.
2016;44:259-70. https://doi.org/10.1111/apt.13665 PMid:27237709 PMCid:PMC5089582
- Aydinok
Y, Evans P, Manz CY, Porter JB. Timed non-transferrin bound iron
determinations probe the origin of chelatable iron pools during
deferiprone regimens and predict chelation response. Haematologica.
2012;97:835-41. https://doi.org/10.3324/haematol.2011.056317 PMid:22180427 PMCid:PMC3366647
- Porter
JB1, Abeysinghe RD, Marshall L, Hider RC, Singh S. Kinetics of removal
and reappearance of non-transferrin-bound plasma iron with deferoxamine
therapy. Blood. 1996;88:705-13. https://doi.org/10.1182/blood.V88.2.705.bloodjournal882705
- Lal
A, Porter J, Sweeters N, Ng V, Evans P, Neumayr L, Kurio G, Harmatz P,
Vichinsky E. Combined chelation therapy with deferasirox and
deferoxamine in thalassemia. Blood Cells Mol Dis. 2013;50:99-104. https://doi.org/10.1016/j.bcmd.2012.10.006 PMid:23151373 PMCid:PMC3592978
- Giardina
PJ, Grady RW. Chelation therapy in beta-thalassemia: the benefits and
limitations of desferrioxamine. Semin Hematol. 1995;32:304-12.
- Miskin
H, Yaniv I, Berant M, Hershko C, Tamary H. Reversal of cardiac
complications in thalassemia major by long-term intermittent daily
intensive iron chelation. Eur J Haematol. 2003;70:398-403. https://doi.org/10.1034/j.1600-0609.2003.00075.x PMid:12756023
- Salvarani
C, Baricchi R, Lasagni D, Boiardi L, Piccinini R, Brunati C, Macchioni
P, Portioli I. Effects of desferrioxamine therapy on chronic disease
anemia associated with rheumatoid arthritis. Rheumatol Int.
1996;16:45-8. https://doi.org/10.1007/BF01816434 PMid:8853224
- Vreugdenhil
G, Swaak AJ, Kontoghiorghes GJ, van Eijk HG. Efficacy and safety of
oral iron chelator L1 in anaemic rheumatoid arthritis patients. Lancet.
1989;2(8676):1398-9. https://doi.org/10.1016/S0140-6736(89)92011-4
- Bruin
GJ, Faller T, Wiegand H, Schweitzer A, Nick H, Schneider J, Boernsen
KO, Waldmeier F. Pharmacokinetics, distribution, metabolism, and
excretion of deferasirox and its iron complex in rats. Drug Metab
Dispos. 2008;36:2523-38. https://doi.org/10.1124/dmd.108.022962 PMid:18775980
- Pippard
MJ, Jackson MJ, Hoffman K, Petrou M, Modell CB. Iron chelation using
subcutaneous infusions of diethylene triamine penta-acetic acid (DTPA).
Scand J Haematol. 1986;36:466-72. https://doi.org/10.1111/j.1600-0609.1986.tb02282.x PMid:3738427
- De
Virgiliis S, Congia M, Turco MP, Frau F, Dessi C, Argiolu F, Sorcinelli
R, Sitzia A, Cao A. Depletion of trace elements and acute ocular
toxicity induced by desferrioxamine in patients with thalassaemia. Arch
Dis Child. 1988;63:250-5. https://doi.org/10.1136/adc.63.3.250 PMid:3355204 PMCid:PMC1778769
- Bartakke
S, Bavdekar SB, Kondurkar P, Muranjan MN, Manglani MV, Sharma R. Effect
of deferiprone on urinary zinc excretion in multiply transfused
children with thalassemia major. Indian Pediatr. 2005;42:150-4.
- al-Refaie
FN, Wonke B, Hoffbrand AV, Wickens DG, Nortey P, Kontoghiorghes GJ.
Efficacy and possible adverse effects of the oral iron chelator
1,2-dimethyl-3-hydroxypyrid-4-one (L1) in thalassemia major. Blood.
1992;80:593-9. https://doi.org/10.1182/blood.V80.3.593.593 PMid:1638018
- Erdoğan
E, Canatan D, Ormeci AR, Vural H, Aylak F. The effects of chelators on
zinc levels in patients with thalassemia major. J Trace Elem Med Biol.
2013;27:109-11. https://doi.org/10.1016/j.jtemb.2012.10.002 PMid:23164519
- Canteros
A, Díaz-Corte C, Fernández-Martín JL, Gago E, Fernández-Merayo C,
Cannata J. Ultrafiltrable aluminium after very low doses of
desferrioxamine. Nephrol Dial Transplant. 1998;13:1538-42. https://doi.org/10.1093/ndt/13.6.1538 PMid:9641189
- Kontoghiorghes
GJ, Barr J, Baillod RA. Studies of aluminium mobilization in renal
dialysis patients using the oral chelator
1,2-dimethyl-3-hydroxypyrid-4-one. Arzneimittelforschung. 1994;44:522-6.
- Singh
S, Mohammed N, Ackerman R, Porter JB, Hider RC. Quantification of
desferrioxamine and its iron chelating metabolites by high-performance
liquid chromatography and simultaneous ultraviolet-visible/radioactive
detection. Anal Biochem. 1992;203:116-20. https://doi.org/10.1016/0003-2697(92)90050-H
- Waldmeier
F, Bruin GJ, Glaenzel U, Hazell K, Sechaud R, Warrington S, Porter JB.
Pharmacokinetics, metabolism, and disposition of deferasirox in
beta-thalassemic patients with transfusion-dependent iron overload who
are at pharmacokinetic steady state. Drug Metab Dispos. 2010;38:808-16.
https://doi.org/10.1124/dmd.109.030833 PMid:20097723
- Hasinoff
BB, Patel D, Wu X. The oral iron chelator ICL670A (deferasirox) does
not protect myocytes against doxorubicin.Free Radic Biol Med.
2003;35:1469-79 https://doi.org/10.1016/j.freeradbiomed.2003.08.005 PMid:14642395
- Elalfy
MS, Adly AM, Wali Y, Tony S, Samir A, Elhenawy YI. Efficacy and safety
of a novel combination of two oral chelators deferasirox/deferiprone
over deferoxamine/deferiprone in severely iron overloaded young beta
thalassemia major patients. Eur J Haematol. 2015;95:411-20. https://doi.org/10.1111/ejh.12507 PMid:25600572
- Song
TS, Hsieh YW, Peng CT, Chen TL, Lee HZ, Chung JG, Hour MJ. Combined
versus monotherapy or concurrent therapy for treatment of thalassaemia.
In Vivo. 2014;28:645-9.
- Eghbali A,
Shokri P, Afzal RR, Bagheri B. A 1-year randomized trial of deferasirox
alone versus deferasirox and deferoxamine combination for the treatment
of iron overload in thalassemia major.Transfus Apher Sci.
2019;58:429-33. https://doi.org/10.1016/j.transci.2019.03.021 PMid:31229401
- Cassinerio
E, Orofino N, Roghi A, Duca L, Poggiali E, Fraquelli M, Zanaboni L,
Cappellini MD. Combination of deferasirox and deferoxamine in clinical
practice: an alternative scheme of chelation in thalassemia major
patients. Blood Cells Mol Dis. 2014;53:164-7. https://doi.org/10.1016/j.bcmd.2014.04.006 PMid:24846580
- Elalfy
MS, Saber MM, Adly AA, Ismail EA, Tarif M, Ibrahim F, Elalfy OM. Role
of vitamin C as an adjuvant therapy to different iron chelators in
young β-thalassemia major patients: efficacy and safety in relation to
tissue iron overload. Eur J Haematol. 2016;96:318-26. https://doi.org/10.1111/ejh.12594 PMid:26018112
- Conte
D, Brunelli L, Ferrario L, Mandelli C, Quatrini M, Velio P, Bianchi PA.
Effect of ascorbic acid on desferrioxamine-induced urinary iron
excretion in idiopathic hemochromatosis. Acta Haematol. 1984;72:117-20.
https://doi.org/10.1159/000206370 PMid:6437113
- Kontoghiorghes
GJ, Jackson MJ, Lunec J. In vitro screening of iron chelators using
models of free radical damage. Free Radic Res Commun. 1986;2:115-24. https://doi.org/10.3109/10715768609088062 PMid:3505236
- Mostert
LJ, Van Dorst JA, Koster JF, Van Eijk HG, Kontoghiorghes GJ. Free
radical and cytotoxic effects of chelators and their iron complexes in
the hepatocyte. Free Radic Res Commun. 1987;3:379-88. https://doi.org/10.3109/10715768709088079 PMid:3508452
- Walter
PB, Macklin EA, Porter J, Evans P, Kwiatkowski JL, Neufeld EJ, Coates
T, Giardina PJ, Vichinsky E, Olivieri N, Alberti D, Holland J, Harmatz
P; Thalassemia Clinical Research Network. Inflammation and
oxidant-stress in beta-thalassemia patients treated with iron chelators
deferasirox (ICL670) or deferoxamine: an ancillary study of the
Novartis CICL670A0107 trial. Haematologica. 2008;93:817-25. https://doi.org/10.3324/haematol.11755 PMid:18469351
[TOP]