Hui
Liu1, Lorraine Gemmell1,
Rui Lin1, Fengrong Zuo1, Henry H. Balfour, Jr.2,
Jennifer C. Woo1 and Gregory M. Hayes1.
1
AstraZeneca, South San Francisco, CA 94080, USA.
2 University of Minnesota Medical School,
Minneapolis, MN 55455, USA.
Corresponding
author: Hui Liu. AstraZeneca, 121 Oyster Point Blvd, South San
Francisco, CA 94080, USA. Tel: 650-379-3076. E-mail:
hui.liu@astrazeneca.com
Published: March 1, 2020
Received: November 15, 2020
Accepted: February 10, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020016 DOI
10.4084/MJHID.2020.016
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
No
licensed vaccine is available for prevention of EBV-associated
diseases, and robust, high-throughput bioanalytical assays are needed
to evaluate immunogenicity of gp350 subunit-based candidate EBV
vaccines. Here we have developed an improved EBV-GFP based
neutralization assay for such a vaccine’s pre-clinical and clinical
validation to measure EBV specific neutralizing antibodies in human
donors. The supplementation of guinea pig complement of our previously
published high-throughput EBV-GFP fluorescent focus (FFA)-based
neutralization assay allowed the detection of complement-dependent
neutralizing antibodies using a panel of heat-inactivated healthy human
sera. Anti-gp350 antibody titers, which were evaluated using a
previously optimized anti-gp350 IgG ELISA assay, were moderately
correlated to the FFA-based neutralization titers. Overall, this
improved high-throughput neutralization assay is capable of
characterizing the serologic neutralizing antibody response to natural
EBV infection, with applications in evaluating EBV antibody status in
epidemiologic studies and immunogenicity of candidate gp350-subunit EBV
vaccines in clinical studies.
|
Introduction
Epstein-Barr
virus (EBV) is a prevalent gamma herpes virus and is the causative
agent of infectious mononucleosis (IM), a clinical syndrome
characterized by fever, pharyngitis, and cervical lymphadenopathy
primarily afflicting adolescents and young adults.[1,2]
The virus is transmitted mainly via saliva and is able to infect naïve
B cells through binding of the major viral surface glycoprotein gp350
to CD21 (also called CR2) on the B-cell surface via gB and gH/gL/gp42,
whereas virus infection of epithelial cells in the absence of CR2 can
utilize gH/gL without gp42.[3]
Following primary
infection in B-cells, the virus establishes latency in B cells where it
persists for life. This chronic B-cell reservoir can undergo recurrent
lytic cycle reactivation, asymptomatic shedding of virus into the
saliva, and spread to uninfected individuals, thus facilitating near
complete permeation of the human population. In addition to IM, EBV has
been associated with a variety of malignant diseases including Burkitt
and Hodgkin lymphomas, nasopharyngeal carcinoma, gastric
adenocarcinoma, and post-transplant lymphoproliferative disorder.[4-7]
Despite the ubiquitous prevalence of EBV and associated acute IM
disease, the immune response to EBV infection is not completely
understood. Of particular interest are the immune correlates associated
with symptomatic EBV infection, or IM, compared with asymptomatic EBV
infection.
At present, no therapeutic or prophylactic options are
approved for the prevention or treatment of EBV-associated diseases.
Within the prophylactic approach, both adjuvanted gp350 subunit and
vaccinia-vectored gp350 approaches have been evaluated in humans based
upon the identification that the majority of neutralizing factors
present in EBV-positive serum that is directed against the viral
surface glycoprotein, gp350.[8-10]
Safety and efficacy
trials have been performed using a CHO-derived soluble gp350 subunit
antigen mixed with 3-O-desacyl-4’-monophosphoryl A (AS04) adjuvant.[11,12]
These studies successfully demonstrated safety, tolerability, and
immunogenicity in young adults, where the vaccine induced strong
antibody responses to gp350. The small phase 2 proof-of-concept trial
also revealed a high level of efficacy at preventing acute IM, reaching
100% protection following the third dose. Although clinical protection
was observed in this study, a limited evaluation of the immune response
was performed.
Historically, EBV neutralizing titers have been
quantified using a B cell transformation assay. This method has low
sensitivity and is time consuming as it traditionally requires a 4—6
week incubation period for B cell transformation followed by a
calculation of neutralizing antibody titers. More recently, the
development of a rapid EBV neutralization assay utilizing Raji cells
has been described.[13] This is
based on infection of
an in vitro human B cell line with an EBV encoding green fluorescent
protein (GFP) allowing for detection of neutralizing antibodies by flow
cytometry. This flow cytometry-based method enables the calculation of
neutralization titers within 48 hours and correlates highly with the
historically utilized transformation assay. Although a significant
improvement on the traditional B cell transformation assay, the flow
cytometry-based platform exhibited low throughput and reduced
sensitivity. An adherent cell line, SVK, expressing the native EBV
receptor CD21 (also called CR2; SVK-CR2),[14]
was utilized for EBV neutralization assay.[15]
To increase sample throughput, a fluorescent focus assay (FFA)-based
EBV micro-neutralization assay was also developed with SVK-CR2 cells.
The report by Lin et al. facilitated our previously described
utilization of IsoCyteTM
instrumentation, an automation-friendly benchtop laser scanning
cytometer, to allow for higher-throughput sample testing.[16]
Based
upon previous studies where guinea pig complement has been shown to
expand the linear range of viral neutralization assays,[17]
the current study demonstrates that the inclusion of guinea pig
complement improves both the flow cytometry-based Raji and FFA-based
SVK-CR2 assays by allowing detection of complement-dependent
neutralizing antibodies. Together these results facilitate the
supplementation of guinea pig complement of the high-throughput EBV-GFP
SVK-CR2 FFA-based and EBV-GFP Raji FACS-based micro-neutralization
assays to determine EBV neutralizing titers in EBV vaccine research and
development.
An anti-gp350 IgG ELISA assay, which was previously
optimized through gp350 coating concentration and sera dilutions and
described by Wu et al.,[18] was
utilized to quantify
the human anti-gp350 antibody titers across a panel of healthy human
donors whose EBV antibody status was pre-determined using commercial
ELISAs.
Materials and Methods
Human
sera panel to assess anti-gp350 antibody and neutralization antibody
titers.
Healthy human donor serum samples were purchased from AllCells, LLC and
Bioreclamation, LLC (n=39). Samples were heated at 56ºC for 30 minutes
to inactivate complement, and then the EBV antibody status was
determined using commercial kits (Diamedix, Miami Lakes, FL) to measure
IgG and IgM antibody responses to EBV viral capsid antigen (VCA). The
panel of sera was then examined with the anti-gp350 IgG ELISA, flow
cytometry (FACS)-based micro-neutralization assay and FFA-based
micro-neutralization assay with or without guinea pig complement
supplement.
Anti-gp350
IgG ELISA.
Immulon 4HBX High-binding 96-well plates (ThermoFisher Scientific) were
coated with 0.25 µg/ml recombinant purified gp350 (suspension CHO cell
line produced)[19] in PBS
overnight at 4ºC
(all subsequent steps were performed at room temperature). The plates
were washed 4 times with PBST (PBS with 0.05% Tween-20) and blocked
with Superblock (ThermoFisher Scientific) for 1 hour. Human serum
samples along with positive and negative control serum were serially
diluted 1:5 in dilution buffer containing 0.1% Superblock in PBST (1:10
to 1:97,656,250 for sample, 1:10 to 1:781,250 for positive control, and
1:10 to 1:1250 for negative control) at 100 µL/well and incubated for 1
hour. After washing, HRP-conjugated secondary antibody (Rabbit
anti-human IgG HRP, Dako# P0214) was diluted to 1: 7,500 in dilution
buffer and incubated for 1 hour, followed by 4 washes in PBST. TMB
substrate (SIGMA) was added at 100 µL/well for colorimetric reading.
After adding 1N HCl at 100µL/well to stop the reaction, the absorption
was measured at OD450 on a SpectraMax plate reader. The antiserum
endpoint titer was quantified as the reciprocal dilution factor using
SoftmaxPro to calculate the 4-fold rise above the ELISA assay
background. The lower limit of detection (LOD) of this assay is 10.
Negative samples were assigned an artificial titer of 0.1 for graphing
purpose.
Cells.
Akata-BX1-g, a lymphoma cell line engineered to express GFP in the EBV
virus genome,[20]
was received from Dr. Lindsey Hutt-Fletcher (Louisiana State
University). Akata-BX1-g Cells were grown in suspension in RPMI 1640
medium supplemented with 10% heat-inactivated FBS,
Penicillin/Streptomycin, L-Glutamine, and 500 µg/mL G418 at a maximum
passage density of 2x106
cells/ml. SVK-CR2, an epithelial cell line that overexpresses CD21,[14]
was received from Dr. Lindsey Hutt-Fletcher (Louisiana State
University) to measure the infectivity of EBV-GFP. SVK-CR2 cells were
grown in high glucose DMEM supplemented with 10% FBS,
Penicillin/Streptomycin, L-Glutamine, 10 ng/mL Cholera Toxin, and 400
µg/mL G418. Raji cells (ATCC) were grown in suspension in RPMI1640
medium supplemented with 10% FBS, Penicillin/Streptomycin, and
L-Glutamine.
EBV-GFP
virus induction, harvest, and titration. The EBV virus was
obtained by induction of virus production from Akata-BX1-g cells.[20] EBV-GFP virus was purified from 4x109 Akata-BX1-g
cells that had been pelleted down and re-suspended at a concentration
of 4×106
per mL in 1 liter of virus growth medium (RPMI 1640 medium supplemented
with 1% heat-inactivated FBS, Penicillin/Streptomycin, and L-Glutamine)
and induced with 60 mg goat anti human-IgG (MP Biomedicals #55049) for
a final concentration of 60 µg/mL for 5 days. On day 2, an additional 1
liter of virus growth medium was added to achieve a final anti
human-IgG concentration of 30 µg/mL. On day 5, supernatants were
harvested by centrifuging at 4000g for 15 minutes to remove cells,
filtering on a 0.8 µm Nalgene MF75 filter unit, and then centrifuging
at 16,000g for 90 minutes to pellet the virus. Viral pellets were
re-suspended in 20 mL RPMI containing 100 µg/mL bacitracin (Sigma),
aliquoted and stored at -80ºC.
EBV-GFP virus was titrated as previously described.[16]
SVK-CR2 cells were seeded in 96 well plates at 104
cells/well one day prior to assaying. Fifty µL of two-fold serially
diluted EBV-GFP virus ranging from 1:20 dilution to 1:1280 dilution was
added and cultured for 24-42 hours in a humidified 37ºC, 5% CO2
incubator. Green fluorescent foci indicative of infected SVK-CR2 cells
in each well were enumerated automatically on an IsoCyte device
(Molecular Devices) as FFU/well. The viral titers were reported as Log10 (FFU/mL) = Log10 (Virus
dilution factor x 20 x FFU/well)
Flow
cytometry (FACS)-based micro-neutralization assay in Raji cells.
EBV neutralizing antibody titers were determined using the method
described by Sashihara et al.[13]
EBV-GFP virus was quantified by titration of infected Raji B cells. A
dilution targeting 10% infection of Raji cells by EBV-GFP was used. The
assay was performed either with or without 1% guinea pig complement
(Lonza) in RPMI complete medium. 25 µL of the diluted virus was added
to each well of 96-well U bottom plates before the addition of an equal
volume of 1:2 serially diluted heat-inactivated serum in triplicates
beginning at 1:20 dilution. Viral and serum dilutions were co-incubated
for 2 hours at 37°C before the addition of 105
Raji cells in 200 µL volume, followed by one-day incubation at 37°C.
Cells were pelleted by centrifugation at 1,200 rpm for 5min and fixed
with 200 µL/well of 4% paraformaldehyde for 5 minutes at room
temperature. Fixed cells were washed, re-suspended in DPBS buffer, and
analyzed by flow cytometry on a Guava instrument (Millipore) to
determine the percentage of GFP positive Raji cells. Results were
analyzed with non-linear regression fit using Graphpad Prism software.
The serum dilution at 50% inhibition of virus infection (IC50) was
quantified as titer, and Log2-transformed
data were compared.
EBV-GFP
FFA-based micro-neutralization assay in SVK-CR2 cells.
SVK-CR2 cells were seeded in 96 well plates one day prior to assay.
EBV-GFP with a titer of 500 FFU per well was pre-incubated with the
serially-diluted monoclonal control antibody (72A1, Rockland Inc.) or
heat-inactivated serum samples in triplicates for 30 minutes, and then
SVK-CR2 cells were infected with these serum/virus mixtures as
described in above section. The assay was performed either with or
without 1% guinea pig complement in SVK-CR2 culture medium.
Neutralization titers were calculated as IC50 value in Log2
(serum dilution factor) of non-linear regression fit in GraphPad Prism.
The maximum infection is normalized to virus only as 100%, and the
minimum infection is normalized to cell only as 0%. IC50 corresponds to
antibody dilution at 50% of (Max – Min infection).
Statistics.
Correlation between FFA-based micro-neutralization assay, FCS-based
micro-neutralization assay, and anti-gp350 IgG ELISA were evaluated by
Pearson test using GraphPad Prism software for statistical analysis.
Results
Quantitation
of human anti-gp350 antibody titers with anti-gp350 IgG ELISA.
A total of 39 serum samples were tested for the presence of anti-gp350
antibody titers using the optimized anti-gp350 IgG ELISA assay.
Quantitative analysis, obtained by determination of gp350 endpoint
dilution titers from this sample set (n=39), ranged from negative (LOD
is 10) to a titer of 25830.5, demonstrating a wide range of antibody
titers across this population. Of the 39 samples, six sera tested
negative for VCA IgM and IgG. Three of these six were also below the
limit of detection for gp350 and were classified as anti-gp350
negative. The remaining three VCA negative sera had positive gp350
titers of 220.6, 445.4, and 612.8. Of the 33 EBV VCA seropositive sera,
two sera were VCA IgM positive and VCA IgG negative, and 31 sera were
VCA IgG positive and VCA IgM negative or “equivocal." No sample tested
positive for both VCA IgG and VCA IgM. The two VCA IgM positive sera
were gp350 IgG negative, which makes sense in that the VCA IgM response
occurs within 7—10 days after onset of symptoms, whereas the gp350 IgG
antibody response takes months to develop.[18]
Evidently, these two individuals were in the early stages of a primary
EBV infection. All 31 VCA IgG positive sera were also gp350 IgG
positive. The gp350 titers ranged from 26.2 to 25830.5, with the median
titer at 991.04.
Quantitation
of EBV-GFP neutralization titers by high-throughput fluorescent focus
assay (FFA) or flow cytometry (FACS)-based neutralization assay.
Two different assay formats were used to measure GFP encoding EBV
neutralizing antibody titers in 39 human sera samples: i) the
FACS-based neutralization assay in Raji cells and ii) the FFA-based
neutralization assay in SVK-CR2 cells. In each assay platform, the
human sera were tested in two conditions, supplemented with or without
1% Guinea Pig Complement.
For the FACS-based assay, the mean
neutralization titer of the 39 samples produced without guinea pig
complement, which was expressed as log2
(IC50), was 3.414 [95% confidence interval (CI), 3.293-3.535]. For the
FACS-based assay supplemented with 1% guinea pig complement, the mean
titer was significantly (P<0.0001) increased to 4.890 (95% CI,
4.433-5.347) (Figure 1A).
For
the FFA-based assay, the mean neutralization titer of the 39 samples
produced without guinea pig complement was 6.599 (95% CI, 5.863-7.335).
For the FFA-based assay supplemented with 1% guinea pig complement, the
mean titer was significantly (P<0.0001) increased to 9.314 (95%
CI,
8.466-10.16) (Figure 1A).
We
also compared the two assay formats supplemented with guinea pig
complement, the mean titer produced by SVK-CR2 format (9.314) was also
significantly (P<0.0001) higher than that produced by Raji
format
(4.890) (Figure 1A).
For
the four conditions, FACS without complement, FACS with complement, FFA
without complement, FFA with complement, the detection rate of
neutralizing antibodies above the limit of detection (3.32 Log2 Neut.
titer) was 12.8%, 69.2%, 87.2%, and 89.7% respectively; the detection
rate two folds above of the limit of detection (4.32 Log2 Neut. titer)
was 2.6%, 61.5%, 71.8%, and 89.7% respectively. The correlation between
neutralization titers from FFA-based assay with complement and
FACS-based assay with complement were established (Pearson r value
=0.5931, P value <0.0001, data not shown). The correlation
between
neutralization titers from FFA-based assay with complement and human
anti-gp350 IgG ELISA titers were established as well (for all
39
samples: Pearson r value=0.5573, P value =0.0002, data not shown; for
32 sera samples tested positive in both assay: Pearson r value =0.5844,
P value =0.0004, Figure
1B).
There was no correlation observed between neutralization titers from
FACS-based assay and human anti-gp350 IgG ELISA tiers (data not shown).
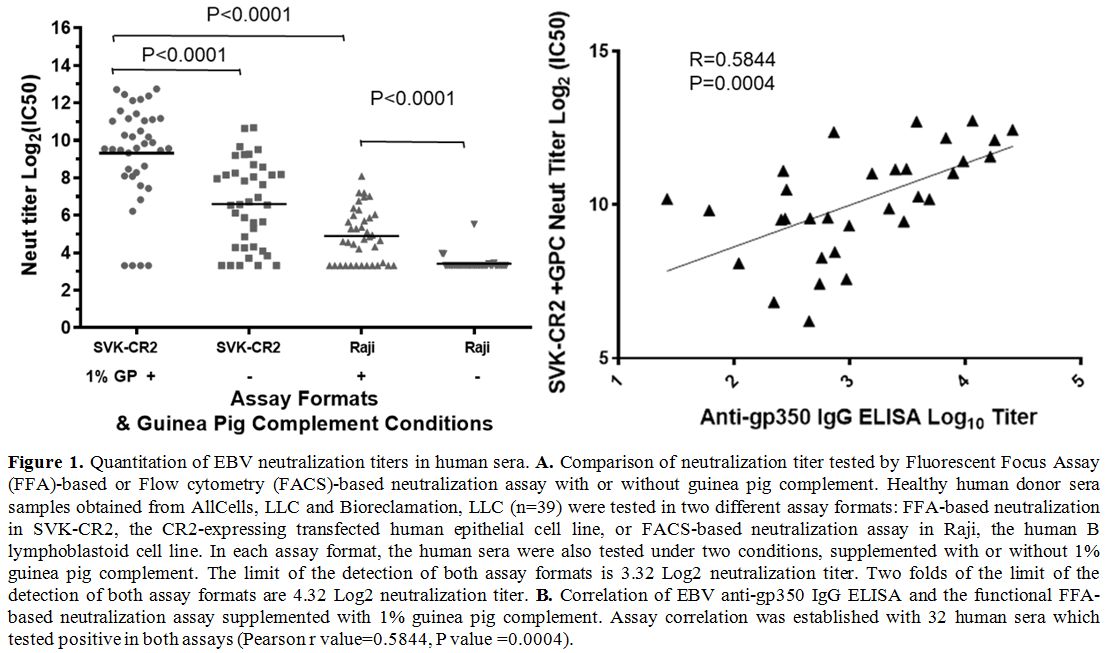 |
Figure
1. Quantitation of EBV neutralization titers in human sera. A.
Comparison of neutralization titer tested by Fluorescent Focus Assay
(FFA)-based or Flow cytometry (FACS)-based neutralization assay with or
without guinea pig complement. Healthy human donor sera samples
obtained from AllCells, LLC and Bioreclamation, LLC (n=39) were tested
in two different assay formats: FFA-based neutralization in SVK-CR2,
the CR2-expressing transfected human epithelial cell line, or
FACS-based neutralization assay in Raji, the human B lymphoblastoid
cell line. In each assay format, the human sera were also tested under
two conditions, supplemented with or without 1% guinea pig complement.
The limit of the detection of both assay formats is 3.32 Log2
neutralization titer. Two folds of the limit of the detection of both
assay formats are 4.32 Log2 neutralization titer. B.
Correlation of EBV anti-gp350 IgG ELISA and the functional FFA-based
neutralization assay supplemented with 1% guinea pig complement. Assay
correlation was established with 32 human sera which tested positive in
both assays (Pearson r value=0.5844, P value =0.0004). |
Discussion
In
this study, an improved guinea pig complement-supplemented
high-throughput EBV-GFP SVK-CR2 FFA-based neutralization assay was
developed for use in clinical investigations of disease outcome
following primary EBV infection. This assay also has utility in staging
the phase of EBV infection, in epidemiologic studies, and in the
clinical development of a prophylactic EBV vaccine or therapeutic
agent. In the determination and assessment of assay parameters suitable
for use in these studies, sera from healthy human donors were used to
confirm the key humoral immune responses against natural EBV infection,
characterized by anti-gp350 IgG titer and EBV neutralizing titer.
Determination
of clinical efficacy for any prophylactic EBV study is predicated upon
the accurate identification and grouping of clinical study participants
into those who have been previously infected by the virus and thus are
EBV seropositive, and those who have not been infected and are EBV
seronegative. The first detectable humoral response to primary EBV
infection is an IgM class antibody titer directed against the viral
capsid antigen (VCA) that is generally found within 7—10 days after
onset of symptoms in >90% of subjects.[21]
All
infected subjects will develop IgG class antibodies to this antigen
within approximately two months of infection and this response will
persist for life.[21] These EBV
specific antibody
responses contrast with the general heterophile antibody response used
in clinics to diagnose EBV infections. Heterophile antibodies are not
directed against EBV proteins but are antibodies that have been
absorbed to guinea pig kidney and that agglutinate mammalian red blood
cells.[18] Based upon the
universality of a
subsequent IgG response to this viral antigen, the detection of
VCA-specific antibodies is most frequently used to determine EBV
infection status (EBV-naïve versus EBV-experienced).
The
determination of a specific response to a prophylactic gp350-based EBV
vaccine is predicated upon the accurate determination of an immune
response directed against EBV gp350 antigen in vaccine recipients. The
most direct approach to evaluating an immune response to the
gp350-based EBV vaccine is to quantify the EBV gp350 antigen-specific
antibodies in human sera samples with ELISA based platform. To achieve
this goal, we previously optimized an anti-gp350 IgG ELISA assay[18]
to quantify the human anti-gp350 antibody titers across a panel of
healthy human donor EBV serum samples. The gp350 ELISA assay methods,
including gp350 coating concentration and sera dilutions, were
optimized with a reference set of EBV seronegative or seropositive sera
to ensure that data generated by this method conformed to the widely
accepted 4-parameter fit ELISA model. The optimized anti-gp350 IgG
ELISA methodology and the identification of positive and negative assay
controls aids in reducing plate-to-plate, temporal, and intra-operator
variability, which enables accurate quantitative determination of
anti-gp350 antibody titers in human sera applicable to clinical trials.
We did observe that three out of six VCA IgG seronegative samples
were gp350 IgG positive. This might suggest that gp350 specific immune
responses detected by our gp350 IgG assay have a lower threshold of
detection compared with the VCA IgG assay or that some samples may be
false positives. This may also represent a reflection of the difference
in timing of EBV VCA vs. gp350 specific response of acute EBV
infection. Therefore, the interpretation of a VCA “seronegative”
results may not exclude EBV infection, and it may be possible to
observe gp350 positive results in these samples as we demonstrate in
this study. Based on our results, an extra set of sera collected at a
later timepoint from VCA seronegative subjects may have value in
confirming their true EBV VCA and anti-gp350 IgG serostatus. Since we
had only a small number of VCA seronegative samples (n=6) in this
study, it may be worth testing more samples in the future to determine
the percentage of anti-gp350 IgG positive samples among the VCA
seronegative samples. If they are found with some frequency and are
true positives based on a panel of other EBV-specific assays,
individuals with this antibody profile would not be considered
appropriate for vaccine trials of EBV-naïve participants.
Lastly,
understanding the mechanism of action of any prophylactic EBV vaccine
is imperative for identifying the correlates of protective immunity to
prevent IM in pre-adolescents and young adults. Human antibodies to EBV
gp350 and EBV gp42 have been shown to block infection of B cells by
EBV. However, anti-EBV gp350 antibodies are reported to neutralize
infectivity more effectively than antibody titers to EBV gp42.[13]
It is also reported that elevated titers of EBV neutralizing antibody
and anti-gp350 antibody were low-risk biomarkers for nasopharyngeal
carcinoma, an EBV-related epithelial tumor.[15]
In this study, a moderate correlation[22]
between anti-gp350 IgG ELISA titer and EBV-GFP SVK-CR2 neutralization
antibody titer in healthy human donor sera was established. Thus, the
induction of high titer EBV gp350 neutralizing antibodies may represent
an essential correlate of protection and mechanism of action to be
monitored.
In this study, two different assay formats, the
FFA-based neutralization assay in SVK-CR2 and the FACS-based
neutralization assay in Raji were compared to measure EBV neutralizing
antibody titers in the same set of 39 healthy human donor sera samples.
The development of a higher throughput FFA-based neutralization assay
was previously published by MedImmune/AstraZeneca.[16]
In our study, we further improved the assay for human sample
application by supplementing an equal amount of the guinea pig
complement to the heat-inactivated human sera. The heat-inactivation
step during the sample processing was to ensure the removal of natural
complement, which might exist in various amounts in human sera and
contribute to the variation of neutralization titer. Currently, there
are two types of neutralizing antibodies that were reported,
complement-independent neutralizing antibodies versus
complement-dependent neutralizing antibodies.[23,24]
Analysis of heat-inactivated sera would get the result for
complement-independent neutralizing antibodies, whereas the addition of
guinea pig complement to heat-inactivated sera would allow the assay to
detect complement-dependent neutralizing antibodies. For the FFA-based
assay, the detection of EBV neutralizing antibodies among the 39
samples increased moderately from 71.8% to 89.7% when the cutoff value
was set as two-fold above limit of detection with the inclusion of
guinea pig complement. For the FACS-based assay, the detection of EBV
neutralizing antibodies increased dramatically from 2.6% to 61.5% with
the inclusion of guinea pig complement. Both assay formats are of
value, as EBV neutralizing analysis using SVK-CR2 cells detects the
titers of neutralizing antibodies against EBV infection of epithelial
cells, whereas neutralizing analysis using Raji cells detects the
titers of neutralizing antibodies against EBV infection of B cells.
Together the results support supplementation of the guinea pig
complement of both high-throughput EBV-GFP SVK-CR2 FFA-based
neutralization assay and EBV-GFP Raji FACS-based assay for
determination of EBV neutralizing titers in human EBV vaccine program.
The throughput of the FFA-based assay also supports its use in large
scale, multicenter studies.
In summary, an improved guinea pig
complement-supplemented high-throughput EBV-GFP SVK-CR2 FFA-based
neutralization assay has been developed for evaluating humoral
responses to EBV during epidemiologic studies, selection, and follow-up
of participants in EBV vaccine trials.
Acknowledgements
We
thank Dr. Lindsey Hutt-Fletcher (Louisiana State University) for
providing Akata-BX1-g cell line and SVK-CR2 cell line. We also thank
Hong Jin (MedImmune/AstraZeneca) for editing the manuscript.
References
- Henle G, Henle W, Diehl
V. Relation of Burkitt's
tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc
Natl Acad Sci U S A 1968; 59:94-101. https://doi.org/10.1073/pnas.59.1.94
PMid:5242134 PMCid:PMC286007
- Balfour HH, Jr.
Progress, prospects, and problems in Epstein-Barr virus vaccine
development. Curr Opin Virol 2014; 6:1-5. https://doi.org/10.1016/j.coviro.2014.02.005
PMid:24632197 PMCid:PMC4072744
- Hutt-Fletcher LM.
Epstein-Barr virus entry. J Virol 2007; 81:7825-32. https://doi.org/10.1128/JVI.00445-07
PMid:17459936 PMCid:PMC1951282
- Andersson
J. An Overview of Epstein-Barr Virus: from Discovery to Future
Directions for Treatment and Prevention. Herpes 2000; 7:76-82.
- Lopes V, Young LS,
Murray PG. Epstein-Barr virus-associated cancers: aetiology and
treatment. Herpes 2003; 10:78-82.
- Nishikawa
J, Iizasa H, Yoshiyama H, Shimokuri K, Kobayashi Y, Sasaki S, Nakamura
M, Yanai H, Sakai K, Suehiro Y, Yamasaki T, Sakaida I. Clinical
Importance of Epstein(-)Barr Virus-Associated Gastric Cancer. Cancers
(Basel) 2018; 10. https://doi.org/10.3390/cancers10060167
PMid:29843478 PMCid:PMC6024931
- Smets
F, Sokal EM. Epstein-Barr virus-related lymphoproliferation in children
after liver transplant: role of immunity, diagnosis, and management.
Pediatr Transplant 2002; 6:280-7. https://doi.org/10.1034/j.1399-3046.2002.02029.x
PMid:12234267
- Hoffman
GJ, Lazarowitz SG, Hayward SD. Monoclonal antibody against a
250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane
antigen and a neutralizing antigen. Proc Natl Acad Sci U S A 1980;
77:2979-83. https://doi.org/10.1073/pnas.77.5.2979
PMid:6248876 PMCid:PMC349530
- Thorley-Lawson
DA, Geilinger K. Monoclonal antibodies against the major glycoprotein
(gp350/220) of Epstein-Barr virus neutralize infectivity. Proc Natl
Acad Sci USA 1980; 77:5307-11. https://doi.org/10.1073/pnas.77.9.5307
PMid:6254073 PMCid:PMC350047
- Thorley-Lawson
DA, Poodry CA. Identification and isolation of the main component
(gp350-gp220) of Epstein-Barr virus responsible for generating
neutralizing antibodies in vivo. J Virol 1982; 43:730-6. https://doi.org/10.1128/JVI.43.2.730-736.1982
PMid:6287039 PMCid:PMC256176
- Moutschen
M, Leonard P, Sokal EM, Smets F, Haumont M, Mazzu P, Bollen A, Denamur
F, Peeters P, Dubin G, Denis M. Phase I/II studies to evaluate safety
and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in
healthy adults. Vaccine 2007; 25:4697-705. https://doi.org/10.1016/j.vaccine.2007.04.008
PMid:17485150
- Sokal
EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels
A, Haumont M, Bollen A, Smets F, Denis M. Recombinant gp350 vaccine for
infectious mononucleosis: a phase 2, randomized, double-blind,
placebo-controlled trial to evaluate the safety, immunogenicity, and
efficacy of an Epstein-Barr virus vaccine in healthy young adults. J
Infect Dis 2007; 196:1749-53. https://doi.org/10.1086/523813
PMid:18190254
- Sashihara
J, Burbelo PD, Savoldo B, Pierson TC, Cohen JI. Human antibody titers
to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of
infectivity better than antibody titers to EBV gp42 using a rapid flow
cytometry-based EBV neutralization assay. Virology 2009; 391:249-56. https://doi.org/10.1016/j.virol.2009.06.013
PMid:19584018 PMCid:PMC2728425
- Li
QX, Young LS, Niedobitek G, Dawson CW, Birkenbach M, Wang F, Rickinson
AB. Epstein-Barr virus infection and replication in a human epithelial
cell system. Nature 1992; 356:347-50. https://doi.org/10.1038/356347a0
PMid:1312681
- Coghill
AE, Bu W, Nguyen H, Hsu WL, Yu KJ, Lou PJ, Wang CP, Chen CJ, Hildesheim
A, Cohen JI. High Levels of Antibody that Neutralize B-cell Infection
of Epstein-Barr Virus and that Bind EBV gp350 Are Associated with a
Lower Risk of Nasopharyngeal Carcinoma. Clin Cancer Res 2016;
22:3451-7. https://doi.org/10.1158/1078-0432.CCR-15-2299
PMid:26920891 PMCid:PMC4947430
- Lin
R, Heeke D, Liu H, Rao E, Marshall JD, Chio V, Cataniag F, Yu L, Zuo F,
McCarthy MP. Development of a robust, higher throughput green
fluorescent protein (GFP)-based Epstein-Barr Virus (EBV)
micro-neutralization assay. J Virol Methods 2017; 247:15-21. https://doi.org/10.1016/j.jviromet.2017.04.012
PMid:28457783
- Elliott
MB, Pryharski KS, Yu Q, Parks CL, Laughlin TS, Gupta CK, Lerch RA,
Randolph VB, LaPierre NA, Dack KM, Hancock GE. Recombinant respiratory
syncytial viruses lacking the C-terminal third of the attachment (G)
protein are immunogenic and attenuated in vivo and in vitro. J Virol
2004; 78:5773-83. https://doi.org/10.1128/JVI.78.11.5773-5783.2004
PMid:15140975 PMCid:PMC415824
- Bu
W, Hayes GM, Liu H, Gemmell L, Schmeling DO, Radecki P, Aguilar F,
Burbelo PD, Woo J, Balfour HH, Jr., Cohen JI. Kinetics of Epstein-Barr
Virus (EBV) Neutralizing and Virus-Specific Antibodies after Primary
Infection with EBV. Clin Vaccine Immunol 2016; 23:363-9. https://doi.org/10.1128/CVI.00674-15
PMid:26888186 PMCid:PMC4820504
- Servat
E, Ro BW, Cayatte C, Gemmell L, Barton C, Rao E, Lin R, Zuo F, Woo JC,
Hayes GM. Identification of the critical attribute(s) of EBV gp350
antigen required for elicitation of a neutralizing antibody response in
vivo. Vaccine 2015; 33:6771-7. https://doi.org/10.1016/j.vaccine.2015.10.024
PMid:26485517
- Molesworth
SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH
is essential for penetration of B cells but also plays a role in
attachment of virus to epithelial cells. J Virol 2000; 74:6324-32. https://doi.org/10.1128/JVI.74.14.6324-6332.2000
PMid:10864642 PMCid:PMC112138
- Odumade
OA, Hogquist KA, Balfour HH, Jr. Progress and problems in understanding
and managing primary Epstein-Barr virus infections. Clin Microbiol Rev
2011; 24:193-209. https://doi.org/10.1128/CMR.00044-10
PMid:21233512 PMCid:PMC3021204
- Mukaka
MM. Statistics corner: A guide to appropriate use of correlation
coefficient in medical research. Malawi Med J 2012; 24:69-71.
- Sairenji
T, Sullivan JL, Humphreys RE. Complement-dependent, Epstein-Barr
virus-neutralizing antibody appearing early in the sera of patients
with infectious mononucleosis. J Infect Dis 1984; 149:763-8. https://doi.org/10.1093/infdis/149.5.763
PMid:6327851
- Sairenji
T, Sullivan JL, Sakamoto K, Spiro RC, Katayama I, Humphreys RE.
Epstein-Barr virus infections in hairy cell leukemia patients in the
presence of complement-dependent neutralizing antibody. Cancer Res
1985; 45:411-5.
[TOP]