Adia E. Adjambri1-2, Sylvie Bouvier3, Roseline N’guessan4, Emma N’draman-Donou1, Mireille Yayo-Ayé1-2, Marie-France Meledje2, Missa L. Adjé2 and Duni Sawadogo1-2.
1 Department of Hematology, Faculty of Pharmacy, Felix Houphouet Boigny University, Abidjan, Côte d’Ivoire.
2 Hematology Unit, Central Laboratory, Yopougon University Hospital, Abidjan, Côte d’Ivoire.
3 Department of Hematology, Nîmes University Hospital, University of Montpellier, France.
4 Department of Pediatrics, Yopougon University Hospital, Abidjan, Côte d’Ivoire.
Corresponding
author: Dr. Adia Eusèbe Adjambri (Pharm D), Department of Hematology,
Faculty of Pharmacy, Felix Houphouet Boigny University, Cocody, P.O.
Box 2308, Abidjan 08, Côte d’Ivoire. Tel: +225 47 40 87 96, Fax:+225 22
41 05 79. E-mail:
eusebeadjambri@yahoo.fr
Published: March 1, 2020
Received: January 1, 2020
Accepted: February 15, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020019 DOI
10.4084/MJHID.2020.019
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Type 3 von Willebrand disease (VWD) is the most severe form of VWD,
characterized by a near-total absence of von Willebrand factor (vWF),
leading to a massive deficiency in plasmatic factor VIII (FVIII). VWD
may be confused with hemophilia A, sometimes leading to misdiagnosis.
The purpose of this work was to finalize the biological diagnosis of
patients with FVIII activity deficiency in Abidjan in order to guide
the best type of management.
Methods:
We conducted a cross-sectional descriptive study from June 2018 to
April 2019. Forty-nine patients, all of whom had lower FVIII levels or
had been referred for a bleeding disorder, were monitored in the
clinical hematology service. The pro-coagulant activity of coagulation
factors was performed in Abidjan. The assays for von Willebrand antigen
and activity were performed at Nîmes University Hospital in France.
Results:
The mean age of patients was 13.8 years (1 – 65) and 86% were Ivorian.
FVIII deficiency was discovered during a biological checkup,
circumcision or post-traumatic bleeding, in 33%, 31% and 29%
respectively. The FVIII deficiency of patients was classified as severe
(89.8%), moderate (8.2%) and mild (2%). Only one patient had a
quantitative deficiency of von Willebrand factor (vWF: Ag <3%) with
undetectable von Willebrand factor activity (vWF: Ac) and an FVIII
level <1%.
Conclusions:
Not all of the congenital deficiency of FVIII are represented by
hemophilia A. It was crucial to assess the Willebrand factor of these
patients followed in Côte d'Ivoire for whom hemophilia A had been
suspected.
|
Introduction
Hemophilia
is an X-linked recessive disease and is the second most common
hereditary hemorrhagic disease after von Willebrand disease (VWD). It
almost exclusively affects males with an average incidence of
approximately one in 5,000 births for hemophilia A (HA) which is
defined by a deficiency in coagulation factor VIII (FVIII), and one in
20,000 to 30,000 births for hemophilia B (HB) which is defined by a
deficiency in factor IX (FIX).[1] These deficiencies result from mutations in the genes encoding for FVIII or FIX.
VWD
is an inherited bleeding disorder caused by a mutation in the gene
encoding the von Willebrand factor (vWF), located on the autosomal
chromosome 12.[2] VWD is classified into 3 main types.
Type 1 VWD is a partial quantitative deficiency, Type 2 is a
qualitative defect subdivided into 4 subtypes (2A, 2B, 2M and 2N) and
Type 3, the least common but most serious form, is a complete
deficiency of Vwf.[3,4] It has an estimated prevalence
of 0.5 to 1 per million in western countries and may be more prevalent
in communities with high consanguinity.[5,6]
FVIII
is chaperoned by the vWF in the blood to protect it from proteolysis,
especially by the activated C protein. Any significant change in vWF is
usually accompanied by a parallel variation in the level of FVIII in
the bloodstream.[7] Therefore, FVIII decrease is found
both in HA and VWD. In Type 3 VWD, the level of FVIII is significantly
reduced, and therefore Type 3 VWD may be confused with severe HA. In
addition to cutaneous and mucous membrane bleeding, which are
characteristic of primary hemostasis disorders, hemarthrosis and
hematomas which are characteristic of coagulation disorder, may also
occur. These features may lead to misdiagnosis.
The aim of this
work was to improve the biological diagnosis of patients with FVIII
deficiency monitored in the clinical hematology department of Yopougon
University Hospital in Abidjan, in order to improve the standard of
care for these patients.
Patients and Methods
Patients.
The 49 patients under study had been referred for therapeutic
management of a hemorrhagic disease associated with a reduction of
FVIII-dependent procoagulant plasma activity, called FVIII: C
deficiency. They came from different families and were monitored in the
clinical hematology department, or referred for coagulation disorder.
All patients had been contacted by telephone to arrange an appointment
to inform them of the study and obtain their written, informed consent.
For children, consent was collected from a family member.
Methods.
This is a cross-sectional descriptive study conducted from June 2018 to
April 2019. Some of the tests, i.e. the coagulation factor assays, were
performed at the central laboratory of Yopougon University Hospital in
Abidjan. The rest (vWF analysis) was performed at the Hematology
Laboratory of Nîmes University Hospital, France. The blood was
collected by the least traumatic venipuncture as possible, on citrated
anticoagulant, 9 volumes to 1. Citrated plasmas poor in platelets were
prepared by double centrifugation at 2 500 rpm for 10 minutes, then
aliquoted and frozen to -80 °C before being shipped to France on dry
ice.
Factor assay.
Factors and activities were measured by a one-stage assay on a
semi-automatic coagulometer by chronometric technique. We used a kit
consisting of human plasma immunodepleted of FVIII and a ready-to-use
APTT reagent and calcium chloride (0.020 mol/l). The factor level was
determined via a calibration line made of calibrated control plasmas.
The admissibility of the assay procedure was validated by control
plasmas.
Phenotype of von Willebrand factor. The vWF analysis was performed on a fully automatic coagulation analyzer.
Functional determination of von Willebrand factor: vWF: Ac.
vWF activity of patient plasma was determined using the reagent, which
uses polystyrene particles coated with a recombinant platelet protein
(Glycoprotein Ib, rGPIb) with two «function gain» mutations allowing
binding of the Willebrand factor in the absence of ristocetin. vWF in
the plasma then spontaneously recognizes rGPIb and induces the
agglutination of polystyrene particles. Agglutination is measured by
turbidimetry.
Von Willebrand factor antigen: vWF: Ag.
Quantitative determination of plasma vWF was carried out by a technique
based on specific polyclonal antibodies. This was, therefore, an assay
for the vWF antigen (vWF: Ag) using an immuno-turbidimetric technique.
Results
Epidemiological and clinical characteristics.
During the study period, the 49 patients included children and adults
aged 4 months to 65 years, with an average age of 13.8 years. The
majority were children (67%, Table 1).
We registered 42 patients of Ivorian nationality (86%). The decrease in
procoagulant factor VIII level was found either during a biological
survey of the family, during circumcision or post-traumatic bleeding in
33%, 31% and 29% of cases, respectively (Table 2). The clinical signs were dominated by the association of hematomas, hemarthrosis, and bleeding from mucous membranes (69%).
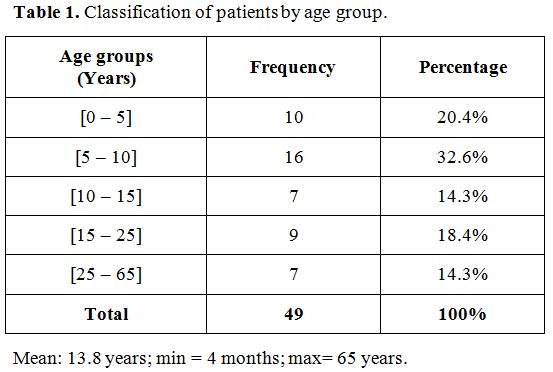 |
Table 1.
Classification of patients by age group. |
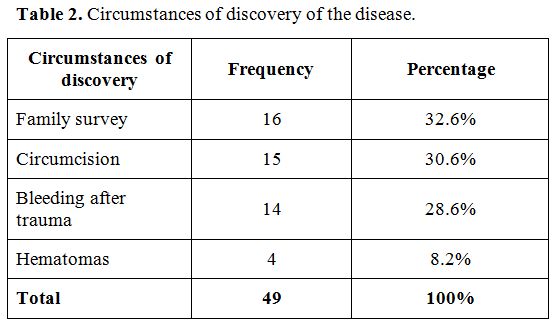 |
Table 2. Circumstances of discovery of the disease. |
Biological data.
In this study, factor VIII deficiency was classified as severe (less
than 1% residual activity), moderate (1 to 5%), and mild (6 to 40%)
respectively in 89.8%, 8.2% and 2% of cases (Table 3).
vWF antigen and activity levels were found to be deficient in one
patient. 71.4% and 87.8% of patients had normal antigen and activity
levels, respectively (Table 4). The patient with vWF deficiency had less than 3% vWF: Ag and an undetectable vWF: Ac, with a FVIII level of less than 1% (Table 5).
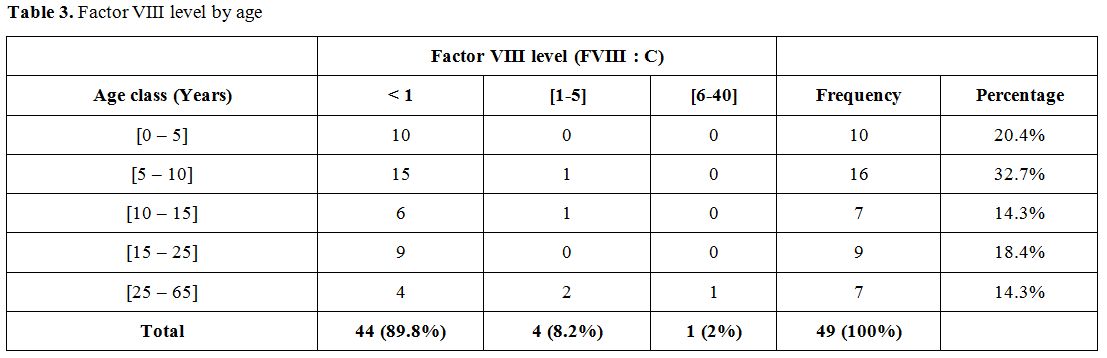 |
Table 3.
Factor VIII level by age |
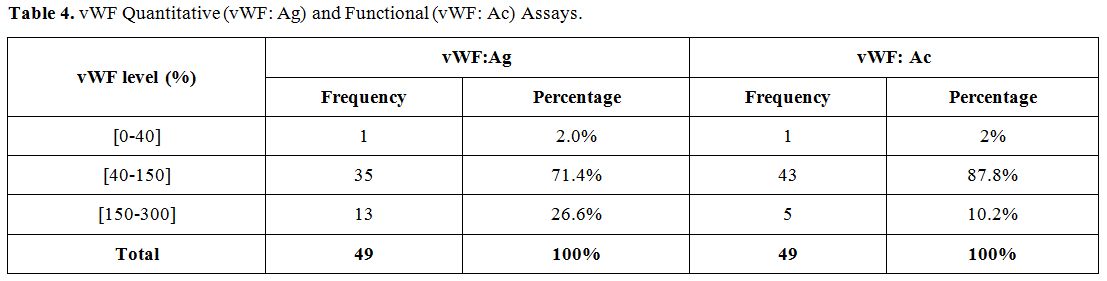 |
Table 4. vWF Quantitative (vWF: Ag) and Functional (vWF: Ac) Assays. |
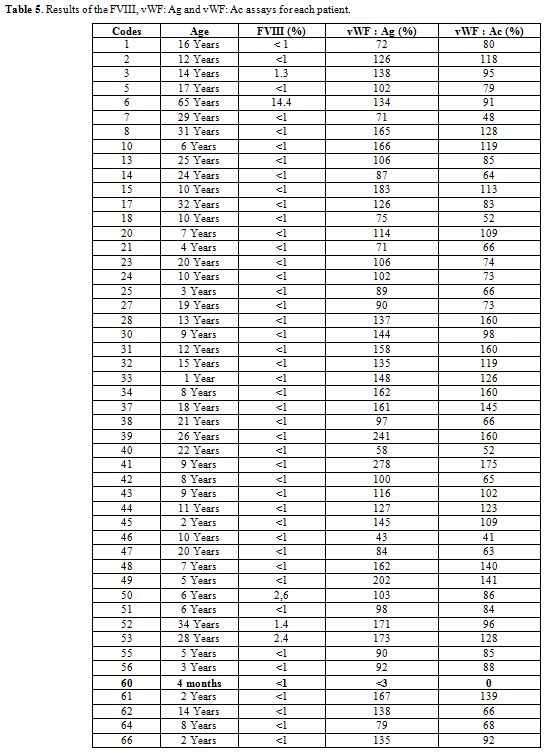 |
Table 5. Results of the FVIII, vWF: Ag and vWF: Ac assays for each patient. |
Discussion
This
study allowed us, for the first time, to evaluate vWF in patients with
low levels of factor VIII (anti-hemophilic factor A), suggesting
hemophilia A. There has been little research on hemophilia and von
Willebrand disease in sub-Saharan African countries,[8,9,10]
particularly in the Côte d’Ivoire. The World Federation of Hemophilia
Report on the Annual Global Survey reported 96 cases of hemophilia in
2018, including 83 hemophiliacs A and 13 hemophiliacs B in our country
with 25,069,229 inhabitants.[11]
More than half
of the study population consisted of children under 15 years of age.
The management of patients with bleeding disorders in our country has
evolved considerably over the last four years, thanks to the World
Federation of Hemophiliacs (WFH). This progress in clinical and
laboratory diagnosis has led to the registration of new patients,
including many children whose disease was discovered following recent
circumcision or a family survey. In the Côte d’Ivoire, circumcision is
practiced during childhood, which explains the high number of children
in our study. Early diagnosis of hemostasis disorders such as
hemophilia and VWD is very important in order to prevent bleeding and
death caused by circumcision and other trauma.[12]
These two causes of bleeding represent 31% and 29% of cases,
respectively. In Africa, circumcision is the most common surgical
procedure for young boys, mainly for religious, cultural and social
reasons.[13]
Severe factor VIII deficiency
largely predominates, 90% compared with just 8% and 2% of moderate and
mild deficiency, respectively. Our results differ from those obtained
by Diop et al. in Senegal, who found a predominance of moderate forms
(56%) versus only 30% of severe forms.[14] Congenital
deficiency in FVIII instinctively leads to hemophilia A. But a plasma
decrease in FVIII can also be observed with abnormality of its carrier
protein, the von Willebrand factor. Evaluation of vWF by antigen assay
and functional activity allowed us to identify one patient with severe
FVIII deficiency associated with absent VWF. This 4-month-old child had
been referred to the department for an isolated prolongation of
activated partial thromboplastin time. This study ultimately allowed us
to diagnose Type 3 von Willebrand disease in the patient. In this
particular case, the hemorrhagic phenotype depended both on the level
of vWF and the level of FVIII.[15,16,17] Several
similar cases of patients with Type 3 von Willebrand disease
misdiagnosed as having hemophilia A have been described in the
literature.[18,19,20,21] The prevalence of Type 3 von
Willebrand disease is very low, ranging from 0.1 to 5.3 per million
inhabitants and varies considerably from one region of the world to
another, with increased prevalence in areas where consanguineous
marriages are more common.[22,23] The highest rate is observed among Arabs and the lowest in southern Europe.[24] The case described in our study is of Arab origin, where consanguineous marriages are common.[25,26,27]
Misdiagnosis
of VWD leads to disparate and inadequate treatment. The therapy in Type
3 VWD aims to correct the combined defects of primary and secondary
hemostasis. This requires restoring a satisfactory level of circulating
vWF which, by stabilizing FVIII, will erase its secondary deficit and
then be accompanied by its reappearance in the plasma. The basic
treatment for Type 3 von Willebrand disease is a substitution treatment
with vWF concentrates of plasmatic or recombinant origin. For example,
if there is time to prevent hemorrhage during a programmed surgery,
these concentrates will induce the delayed reappearance of FVIII. If
urgent treatment is required (for example crucial to treat acute
hemorrhage), then vWF and FVIII should be given. Type 3 von Willebrand
disease patients do not respond to desmopressin,[28,29,30] and this treatment is not recommended. However, there are some discordant data about this therapeutic option.[16]
Conclusions
Our
work highlights the importance of evaluating vWF in patients diagnosed
with factor VIII deficiency and for whom hemophilia A is suspected. Not
all FVIII deficits, however severe, are hemophilic A. The clinical
relevance of the treatment depends on complete phenotyping.
Acknowledgements
The
authors wish to thank all the staff at the hematology laboratory at
Nîmes University Hospital for their technical diagnostic support in
this study. More notably, we wish to thank the hemostasis division who
actively contributed to the study. Thanks also to the association of
hemophiliacs of Côte d'Ivoire (Ivory Coast) and all the patients who
finally agreed to participate in the study.
We thank Teresa
Sawyers, Dr. Yapo Vincent de Paul for expert editorial assistance and
professor Jean-Christophe Gris (Head of the hematology laboratory,
Nîmes University Hospital) for his availability and direction of this
work.
References
- Mannucci PM, Tuddenham EG. The hemophilias - from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779. https://doi.org/10.1056/NEJM200106073442307 PMid:11396445
- Castaman
G and Linari S. Diagnosis and Treatment of von Willebrand Disease and
Rare Bleeding Disorders. J. Clin. Med. 2017; 6(4). pii: E45. https://doi.org/10.3390/jcm6040045 PMid:28394285 PMCid:PMC5406777
- Lehner
S, Ekhlasi-Hundrieser M, Detering C, Allerkamp H, Pfarrer C, von Depka
Prondzinski M. A 12.3-kb Duplication Within the VWF Gene in Pigs
Affected by Von Willebrand Disease Type 3. G3 (Bethesda). 2018; 8(2):
577-585. https://doi.org/10.1534/g3.117.300432 PMid:29208651 PMCid:PMC5919753
- Sadler
JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev
J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL,
Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM,
Srivastava A, Montgomery RR, Federici AB. Update on the pathology and
classification of von Willebrand disease: a report of the subcommittee
on von Willebrand Factor. J Thromb Haemost 2006; 4(10): 2103-2114. https://doi.org/10.1111/j.1538-7836.2006.02146.x PMid:16889557
- Srivastava
A, Rodeghiero F. Epidemiology of von Willebrand disease in developing
countries. Semin Thromb Hemost. 2005; 31(5): 569‐576. https://doi.org/10.1055/s-2005-922229 PMid:16276466
- Elayaperumal
S, Fouzia NA, Biswas A, Nair SC, Viswabandya A, George B, Abraham A,
Oldenburg J, Edison ES, Srivastava A. Type-3 von Willebrand disease in
India-Clinical spectrum and molecular profile. Haemophilia. 2018;
24(6): 930-940. https://doi.org/10.1111/hae.13542 PMid:29984440
- Qachouh M., Harif M., Benchekroun S. La maladie de willebrand. Journal Marocain des Sciences Médicales 2009; 16(3): 7-11. https://revues.imist.ma/index.php?journal=JMSM&page=article&op=view&path%5B%5D=639&path%5B%5D=523
- Gillham
A, Greyling B, Wessels TM, Mbele B, Schwyzer R, Krause A, Mahlangu J.
Uptake of genetic counseling, knowledge of bleeding risks and
psychosocial impact in a south African cohort of female relatives of
people with hemophilia. J Genet Couns. 2015; 24(6): 978-86. https://doi.org/10.1007/s10897-015-9834-8 PMid:25828422
- Naicker T, Aldous C, Thejpal R. Haemophilia: a disease of women as well. S. Afr. J. Child Health. 2016; 10(1): 29-32. https://doi.org/10.1002/cbl.30156
- Seck
M, Faye BF, Sall A, Sy D, Touré SA, Dieng N, Guéye YB, Gadji M, Touré
AO, Costa C, Lasne D, Rothschild C, Diop S. Bleeding risk assessment in
hemophilia a carriers from Dakar, Senegal. Blood Coagul Fibrinolysis.
2017; 28(8): 642-645. https://doi.org/10.1097/MBC.0000000000000653 PMid:28731872
- World federation of hemophilia. Population statistics. In : Report on the Annual Global Survey 2018. 20th Anniversary, Montreal, WFH. 2019; 33-36.
- Lambert
C, Meité N, Sanogo I, Lobet S, Adjambri E, Eeckhoudt S, Hermans C.
Haemophilia in Côte d'Ivoire (Ivory Coast) in 2017: extensives data
collection as part of the World Federation of Haemophilia's twnning
program. Haemophilia. 2019;00:1-8. https://doi.org/10.1111/hae.13682 PMid:30748057
- Seck
M, Sagna A, Guéye MS, Faye BF, Sy D, Touré SA, Sall A, Touré AO, Diop
S. Circumcision in haemophilia using low quantity of factor
concentrates,: experience from Dakar, Senegal. BMC Hematol.
2017;17(1):4-9. https://doi.org/10.1186/s12878-017-0080-1 PMid:28451435 PMCid:PMC5402675
- Diop
S., Touré AO., Thiam D, Dièye M, Diakhaté L. Profil évolutif de
l'hémophilie A au Sénégal: étude prospective réalisée chez 54 patients.
Transfus Clin Biol. 2003; 10(1): 37-40. https://doi.org/10.1016/S1246-7820(02)00002-2
- Zhang
ZP, Lindstedt M, Falk G, Blombäck M, Egberg N, Anvret M. Nonsense
mutations of the von Willebrand factor gene in patients with von
Willebrand disease type III and type I. Am J Hum Genet. 1992; 51(4):
850-858. https://www.ncbi.nlm.nih.gov/pubmed/1415226
- Castaman
G, Lattuada A, Mannucci PM, Rodeghiero F. Factor VIII:C increases after
desmopressin in a subgroup of patients with autosomal recessive severe
von Willebrand disease. Br J Haematol 1995; 89(1): 147-151. https://doi.org/10.1111/j.1365-2141.1995.tb08921.x PMid:7833254
- de
Wee EM, Sanders YV, Mauser-Bunschoten EP, van der Bom JG,
Degenaar-Dujardin ME, Eikenboom J, de Goede-Bolder A, Laros-van Gorkom
BA, Meijer K, Hamulyák K, Nijziel MR, Fijnvandraat K, Leebeek FW.
Determinants of bleeding phenotype in adult patients with moderate or
severe von Willebrand disease. Thromb Haemost 2012; 108(4): 683-692. https://doi.org/10.1160/TH12-04-0244 PMid:22918553
- Lee
AC, Li CH, Wong LG : A case of von Willebrand disease type 3
misdiagnosed as hemophilia A. Zhonghua Er Ke Za Zhi 2007; 45(12): 950.
- Boylan
B, Rice AS, De Staercke C, Eyster ME, Yaish HM, Knoll CM, Bean CJ,
Miller CH. Evaluation of von Willebrand factor phenotypes and genotypes
in Hemophilia A patients with and without identified F8 mutations. J
Thromb Haemost 2015; 13(6): 1036-1042. https://doi.org/10.1111/jth.12902 PMid:25780857 PMCid:PMC4512234
- Echahdi
H, El Hasbaoui B, El Khorassani M, Agadr A, Khattab M. Von Willebrand's
disease: case report and review of literature. Pan Afr Med J. 2017; 27:
147. https://doi.org/10.11604/pamj.2017.27.147.12248 PMid:28904675 PMCid:PMC5567960
- Benlaldj
D, Moueden MA, Seghier F. Maladie de von Willebrand type 3 faussement
diagnostiquée en hémophilie A modérée: à propos d'une observation. Rev
Med Brux 2017; 38(1): 36-38.
- Eikenboom
JC. Congenital von Willebrand disease type 3: Clinical manifestations,
pathophysiology and molecular biology. Best Pract Res Clin Haematol.
2001; 14(2) :365-79. https://doi.org/10.1053/beha.2001.0139 PMid:11686105
- Bowman
M, Tuttle A, Notley C, Brown C, Tinlin S, Deforest M, Leggo J,
Blanchette VS, Lillicrap D, James P. The Genetics of Canadian Type 3
von Willebrand Disease (VWD): Further Evidence for Co-dominant
Inheritance of Mutant Alleles. J Thromb Haemost. 2013; 11(3): 512-520. https://doi.org/10.1111/jth.12130 PMid:23311757 PMCid:PMC3904644
- Mannucci
PM, Bloom AL, Larrieu MJ Nilsson IM, West RR. Atherosclerosis and von
Willebrand factor. I. Prevalence of severe von Willebrand's disease in
western Europe and Israel. Br J Haematol. 1984; 57(1) : 163-169. https://doi.org/10.1111/j.1365-2141.1984.tb02876.x PMid:6609712
- Hussain,
R, Bittles AH. The prevalence and demographic characteristics of
consanguineous marriages in Pakistan. J Biosoc Sci 1998, 30 (2):
261-275. https://doi.org/10.1017/S0021932098002612 PMid:9746828
- Denic, S. Consanguinity as risk factor for cervical carcinoma. Med Hypotheses 2003; 60 (3): 321-324. https://doi.org/10.1016/S0306-9877(02)00389-4
- Talbi
J, Khadmaoui AE, Soulaymani AEM, Chafik AEA. Etude de la consanguinité
dans la population marocaine. Impact sur le profil de la santé.
Antropo. 2007, 15: 1-11.
- Castaman G,
Federici AB, Rodeghiero F, Mannucci PM: Von Willebrand's disease in the
year 2003: towards the complete identification of gene defects for
correct diagnosis and treatment. Haematologica 2003; 88(1) : 94-108.
- Federici
AB, Castaman G, Franchini M. Clinical use of Haemate P in inherited von
Willebrand's disease: a cohort study on 100 Italian patients.
Haematologica 2007; 92(7): 944-951. https://doi.org/10.3324/haematol.11124 PMid:17606445
- Federici
AB, James P. Current Management of Patients with Severe von Willebrand
Disease Type 3: A 2012 Update. Acta Haematol. 2012; 128(2): 88-99. https://doi.org/10.1159/000338208 PMid:22722677
[TOP]