Kun Yang1,2, Yi Wu2,3, Yali Zhou2, Binbin Long4, Qian Lu5, Tianhong Zhou2, Li Wang2, Zhili Geng2 and Xiaolin Yin2.
1 Graduate School of Guangxi University of Chinese Medicine, Nanning, China.
2 Department of Hematology, The 923rd Hospital of the Joint Logistics Support Force of the Peoples Liberation Army, Nanning, China.
3 Graduate School of Guilin Medical University, Guilin, China.
4 People's Hospital of Guiping, Guiping, China.
5 People's Hospital of Hezhou, Hezhou, China.
Corresponding
author: Dr. Xiaolin Yin, Department of Hematology, The 923
rd Hospital
of the Joint Logistics Support Force of the Peoples Liberation Army,
Nanning, Guangxi, China; E-mail:
yin-xl@163.com
Published: May 1, 2020
Received: November 28, 2019
Accepted: March 3, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020021 DOI
10.4084/MJHID.2020.021
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Objective: This study focused on the efficacy and safety of thalidomide for patients with β-thalassemia in a multicenter trial.
Methods:
Patients with non-transfusion-dependent thalassemia (NTDT) or
transfusion-dependent thalassemia (TDT), who were unable to pursue
conventional therapy with transfusion and chelation, were recruited
over 3 years in three centers in southern China. We evaluated the
efficacy and safety of thalidomide in the short-term (three months) and
long-term follow-up (12 and 24 months). Response to thalidomide was
defined as follows: Main Responder (MaR) showing an increase in
hemoglobin (Hb) level of >2.0 g/dl or free from blood transfusion
and Minor Responder (MiR) achieving elevated Hb level of 1.0-2.0 g/dl
or ≥50% reduction in blood transfusion frequency.
Results:
The overall response rate (ORR) was 93.5%, with MaR and MiR rates
accounting for 62.9% and 30.6% in short-term follow-up. For patients
with NTDT, the Hb level increased from a baseline mean of 6.8±1.1 g/dl
to 9.7±1.9 g/dl (P<0.001).
Elevated Hb was mainly attributable to increased fetal hemoglobin (HbF)
levels. Among patients with TDT, while an increase in the average Hb
concentration was observed, there was a significant drop in yearly
transfusions from 20.7±7.7 to 5.8±6.8 blood units per year (P<0.001).
The response of patients in both categories was sustained even after an
average follow up of 14.6±9.6 months (3-37 months). Minimal
side-effects were documented throughout, except peripheral
neurotoxicity in one patient. Logistic regression analysis identified
the ratio of HbF at baseline (P=0.038, OR=1.111, 95% CI: 1.006-1.226) as an independent risk factor for the primary response to thalidomide.
Conclusion:
Thalidomide had significant therapeutic effects on patients with
β-thalassemia with a sustained response. Peripheral neuropathy is one
of the most feared complications. While these preliminary results
support the potential long-term efficacy of thalidomide as a
therapeutic agent for β-thalassemia, several issues need to be
addressed before its application in the clinic.
|
Introduction
Thalassemia
incorporates a group of hereditary hematological diseases caused by
disorders in α/β-globin chain synthesis. Currently, thalassemia
syndromes can be classified phenotypically into
non-transfusion-dependent thalassemia (NTDT) or transfusion-dependent
thalassemia (TDT) based on their clinical severity and transfusion
requirements.[1] However, more transfusions may also
be required for NTDT patients whose clinical course has evolved with
age. Such patients ultimately become regularly transfused.[2,3] The predominant hemoglobin (Hb) variant expressed by the fetus and newborn is fetal hemoglobin (HbF, α2γ2), is progressively replaced with adult hemoglobin (HbA, α2β2)
after birth. In patients with β-thalassemia, γ-globin can combine with
redundant α-globin chains and compensate for the lack of β-globin
chains.[1] Observational studies have suggested that
inducing synthesis of HbF may be effective in alleviating clinical
manifestations in β-thalassemia patients.[4,5] Drugs capable of improving the synthesis of HbF and then anemia and quality of life have therefore been investigated.
Several promising fetal hemoglobin (HbF) inducers, including hydroxyurea, erythropoietin, 5-azacytidine, and sodium butyrate,[4,6]
have achieved limited success for the treatment of β-thalassemia to
date. Hydroxyurea, approved by the FDA for the treatment of sickle cell
disease (SCD), is also the most widely used HbF inducer in
β-thalassemia. However, the clinical application of hydroxyurea is
limited by the low number of significant responders,[7,8] reduced clinical response with long follow-up,[9,10] and bone marrow suppression.[11]
Therefore, better treatments facilitating improved outcomes are
increasingly important. Thalidomide, a synthetic glutamic acid
derivative, is widely used as an immunomodulator for the treatment of
various hematological cancers due to its anti-inflammatory,
anti-angiogenic, and anti-tumor effects.[12] Moreover, thalidomide is an HbF inducer that promotes γ-globin gene expression.[13,14] A few case reports and retrospective analyses have documented significant effects of thalidomide on NTDT or TDT,[15-20] that our group subsequently confirmed in a clinical trial.[21]
However, the reliability of these studies was softened by the few
patients studied and short-term follow-up. In the current study, we
analyzed the efficacy and safety of thalidomide for β-thalassemia in a
relatively large patient sample over a long-term follow-up period.
Patients and Methods
In
the period from May 2016 to June 2019, 71 patients with duration of
therapy over 3 months, and follow-up data were recruited. The following
inclusion criteria were adopted in the trial: 1) patients with a
clinical and genetic diagnosis of β-thalassemia requiring blood
transfusion, but unable to afford regular transfusions or iron
chelation due to economic or other reasons; 2) patients between 14 and
65 years of age; 3) gender not limited; and 4) an ECOG physical score
between 0 and 2 points. Patients with liver, renal, cardiac, pulmonary,
or neurological deficits were excluded, as were patients with a history
of thrombotic episodes. All females were checked for pregnancy, and
pregnant patients were ruled out. All women enrolled were informed that
they should absolutely avoid pregnancy during treatment and until 6
months after the withdrawal of medicine. Patients were informed of the
side effects and possible benefits of thalidomide. Full informed
consent was required before treatment was initiated. All patients were
followed up by the hematology department of each research center during
the observation period and received thalidomide treatment for at least
three months. Except for supportive care with transfusions and iron
chelation therapy, patients were required not to have received any
therapy that affects Hbs for at least 3 months before starting the
thalidomide treatment. The thalidomide protocol for patients with
β-thalassemia was approved by the Medical Ethics Committee of the 923rd
Hospital of the Joint Logistics Support Force of the Peoples Liberation
Army, People's Hospital of Guiping and People's Hospital of Hezhou. The
clinical trial was registered at ClinicalTrials.gov, registration
number: NCT02995707.
The initial dose of thalidomide used was 50
mg/d, and a daily dose of 100 mg/d was given to patients needing blood
transfusions at least twice a month. Aspirin (100 mg/d) was prescribed
to patients post-splenectomy or those with platelet counts >500 × 109/L
to prevent thrombosis. Patients were regularly followed up monthly
during the first three months of treatment and every 2-3 months
afterward. Baseline and follow-up records were reviewed for demographic
data, transfusion history, splenic size, adverse reaction, and duration
of therapy. A halving of the dosage was prescribed in cases where side
effects were graded III or above. For patients who failed to respond,
50% of the current dose was increased every month after 3 months of
treatment. If no response was observed within 6 months, thalidomide was
discontinued, and the patient advised to resume conventional
management. Within the four research centers, complete blood counts
were analyzed using an XE 5000 automatic blood cell analyzer (Sysmex
Corporation, Kobe, Japan). Different Hb levels were quantified using
Bio-Rad Variant II high-pressure liquid chromatography (HPLC) (Bio-Rad,
Hercules, CA, USA). Biochemical parameters and serum ferritin levels
(SF) were assessed using a multichannel analyzer (Abbot Aeroset, Abbott
Diagnostics, Bohemia, NY, USA) and chemiluminescence (Beckman Coulter,
Inc., CA, USA).
Standard for determination of efficacy.
For patients with NTDT, response to thalidomide was defined as follows:
Main Responder (MaR) showing an increase in Hb level >2.0g/dl, Minor
Responder (MiR) achieving elevation in Hb level of 1.0-2.0g/dl, and No
Responder (NR) showing a <1g/dl increase in Hb level. For patients
with TDT, the groups were defined by the following parameters: MaR,
removal from the blood transfusion, MiR, ≥50% reduction in transfusion
requirement, and NR, <50% reduction in transfusion requirement.[4,7]
Statistical analysis.
SPSS Statistics 21.0 (SPSS Inc., Chicago, IL, USA) was applied for data
analysis. Numerical data were presented as means ± SD or median and
interquartile range (IQR). A paired t-test
or Mann-Wilcoxon rank-sum test was applied to compare the changes in
continual variables before and after treatment. Comparisons in
numerical variables between two groups were performed with Student’s t-test
or Mann-Whitney rank-sum test. Hb levels were assessed for
comparability at each time-point using repeated measures analysis of
variance (ANOVA). Chi-square or Fisher exact test was used to compare
categorical variables for small sample size, followed by logistic
regression using significant results from univariate analysis to
confirm the association. P values <0.05 were considered statistically significant.
Results
Of
the enrolled 71 patients, six were treated for less than 3 months, and
three were without any follow-up data after treatment, leading to the
final inclusion of 62 patients. The patient group comprised 27 males
and 35 females, 39 NTDT and 23 TDT patients, and 29 splenectomized and
33 nonsplenectomized patients. The average age of patients was 27.2±7.9
years (range, 15–45 years). During the treatment period, the initial
dose of 50 mg and 100 mg were 58 patients and 4 patients. Moreover, the
median dose of thalidomide at the last follow-up was 50 mg/d (range
12.5 mg/d–150 mg/d). Fourteen of the patients discontinued treatment,
four of whom were NRs and stopped within 3–6 months, and seven patients
experienced dose modifications. The average duration of thalidomide
treatment was 14.6±9.6 months (range, 3–37 months), with 34 patients
treated over 12 months and 11 over 24 months.
Clinical features.
In our cohort, alleviation of fatigue, and an increase in the energy
state, well-being, and physical activity was detected in 88.7% (55/62)
of the patients. Facial changes were observed in 69.4% (43/62) of the
patients at the end of the study. In nonsplenectomized responders,
average spleen size (length × width) was not significantly altered
(110.1±20.3 cm2 vs 114.7±29.6 cm2, n=14, P=0.493).
Short-term follow-up.
After a 3-month treatment period, 62.9% (39/62) and 30.6% (19/62) of
the patients showed MaR and MiR status, respectively, while 6.5% (4/62)
were classified as NR. As shown in Table 1, for patients with NTDT, the Hb level increased from a baseline mean of 6.8±1.1 g/dl to 9.7±1.9 g/dl (P<0.001),
with an average increase of 2.9±1.6g/dl. Elevated Hb was mainly
attributable to increased HbF levels. The average HbF percentage
increased from a pretreatment level of 41.9±23.4% to 54.3±23.0%
(P<0.001) after treatment. Among patients with TDT, transfusions
were terminated in 43.5% (10/23) of the patients and decreased by more
than 50% in 52.2% (12/23) of the patients, an increase in the average
hemoglobin concentration was contemporarily observed. There was a
significant drop in yearly transfusions from 20.7±7.7 to 5.8±6.8 blood
units per year. After treatment, the red blood cell (RBC) count was
markedly increased, nucleated red blood cells (NRBC) were significantly
decreased, and reticulocyte counts were not significantly changed. As
mean cell volume (MCV) decreased, mean corpuscular Hb concentration
(MCHC) significantly increased. Parameters reflecting hemolysis,
including bilirubin and lactate dehydrogenase (LDH), showed a
significant decrease. However, after the treatment, the overall average
SF was not decreased but increased to a significant extent.
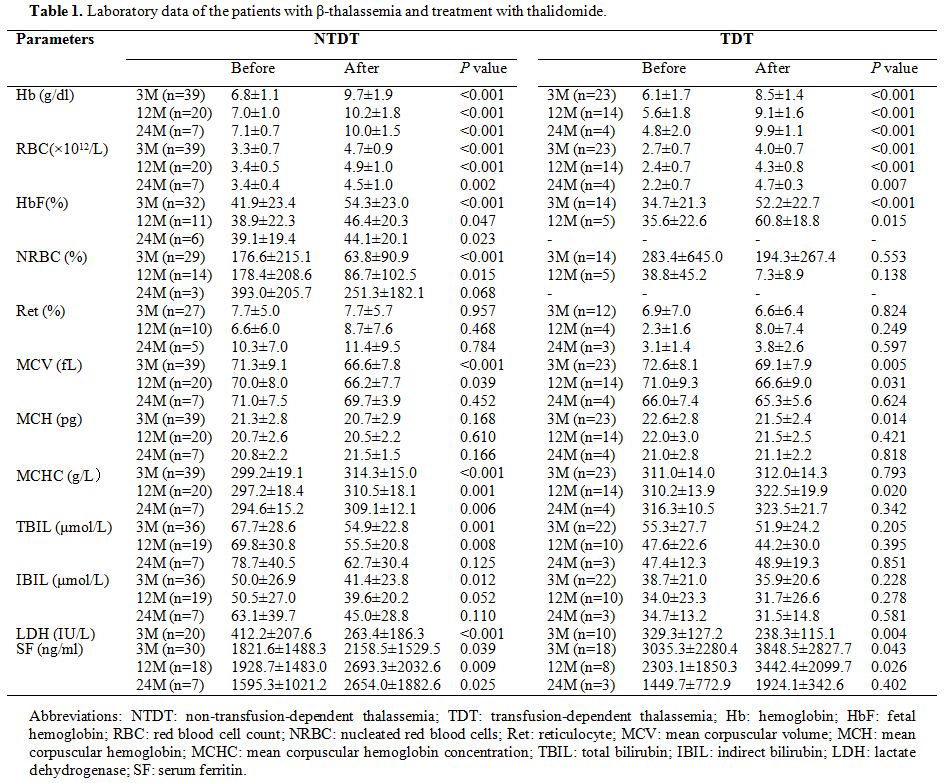 |
Table
1. Laboratory data of the patients with β-thalassemia and treatment with thalidomide. |
Long-term follow-up. Four NRs among the 62 patients discontinued therapy, respectively, after 3-6 months. Table 1
depicts the changes in clinical and laboratory efficacy indicators of
patients with NTDT or TDT after long-term follow-up. In total, 34 of
the 58 responders were treated with thalidomide for >12 months,
averaging 21.9±6.7 months (12-37 months). In 28 responders, the
long-term follow-up response was not significantly different from the
3-month response. Among the remaining six patients, two showed a
decreased therapeutic effect, changing from MaR to MiR status, and four
improved from MiR to MaR. Repeated measures ANOVA showed that Hb levels
at different follow-up time points of 3 and 12 months after treatment
increased significantly, compared with baseline values in NTDT
(F=58.682, P<0.001) and TDT (F=22.259, P<0.001), with no significant differences between the 3 and 12-month period (P>0.05) (Figure 1).
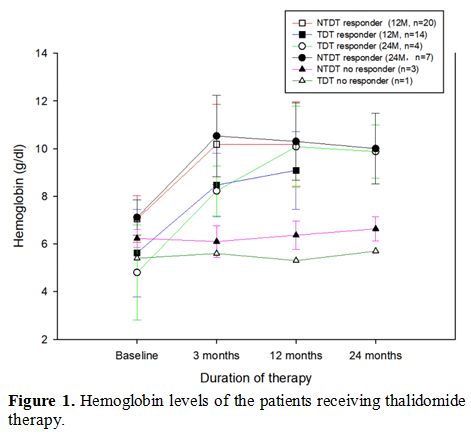 |
Figure 1. Hemoglobin levels of the patients receiving thalidomide therapy. |
The
duration of therapy for 11 patients exceeded 24 months, with an average
treatment period of 30.1±3.8 months (range 24-37 months). Ten patients
maintained clinical response during the observation period, and only
one changed status from MaR to MiR. Similarly, Hb levels of the groups
at different follow-up time-points (3, 12, and 24 months) were
significantly increased compared with baseline levels (NTDT: F=29.411, P<0.001; TDT: F=15.835, P=0.001). Average Hb was comparable at 3, 12, and 24 months (P>0.05) (Figure 1).
Effect of dose adjustment on Hb.
In total, seven patients experienced dose reduction during treatment.
The levels of Hb before and after reduction are shown in Figure 2a.
Two patients with doses reduced from 100 to 50 mg/d maintained constant
levels of Hb. The Hb level decreased in the remaining four patients for
whom the dose was reduced from 50 to 25 mg/d, but three retained MaR
status, and only one changed to MiR.
Interestingly, one patient
required multiple dose adjustments from the initial 50 to 25 mg/d,
which was subsequently reduced to 12.5 mg/d, and finally, 12.5 mg/q.d.,
with the maintenance of Hb level at >9 g/dl, and while the Hb levels
decreased after drug withdrawal, it was restored after resuming
treatment (Figure 2c). Only two
patients were administered increased doses during treatment. In order
to no deviate from blood transfusion, one patient increased the dose
from 50 to 100 mg/d and reached MiR; no improvement was observed in
another patient, even upon increasing the dose from 100 to 150 mg/d.
Effect of drug withdrawal on Hb.
Treatment was discontinued in 14 patients (seven MaR, three MiR, and
four NR). Within the responders, two patients discontinued the drug due
to constipation and menstruation disorders while the others
discontinued treatment because they could not insist on drug intake or
were preparing for conception. The changes in Hb in 10 responders
before and after drug withdrawal are presented in Figure 2b.
Regardless of the length of treatment, the Hb level was significantly
decreased after drug discontinuation for one month and further declined
to baseline levels with the extension of withdrawal time.
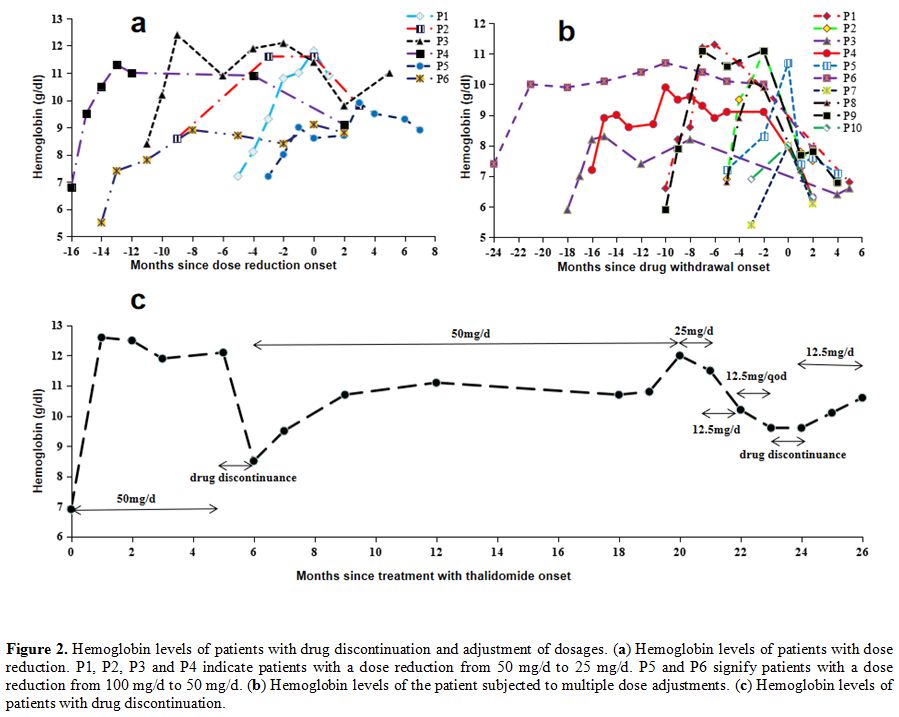 |
Figure 2. Hemoglobin levels of patients with drug discontinuation and adjustment of dosages. (a)
Hemoglobin levels of patients with dose reduction. P1, P2, P3 and P4
indicate patients with a dose reduction from 50 mg/d to 25 mg/d. P5 and
P6 signify patients with a dose reduction from 100 mg/d to 50 mg/d. (b) Hemoglobin levels of the patient subjected to multiple dose adjustments. (c) Hemoglobin levels of patients with drug discontinuation. |
Predictors of response.
To identify potential predictors of the thalidomide response, we
divided all patients into two groups (MaR and MiR+NR). The primary
response to thalidomide was significantly correlated with the HbF ratio
before treatment (P=0.003) and splenic status (P=0.025), but not
related to age, sex, phenotype, duration of treatment, thalidomide dose
or baseline Hb level (Table 2).
To view the effect of blood transfusion on hemoglobin, the authors
analyzed patients with NTDT, separately. Logistic regression analysis
identified the ratio of HbF at baseline (P=0.038, OR=1.111, 95% CI: 1.006-1.226) as an
independent risk factor for the main response to thalidomide.
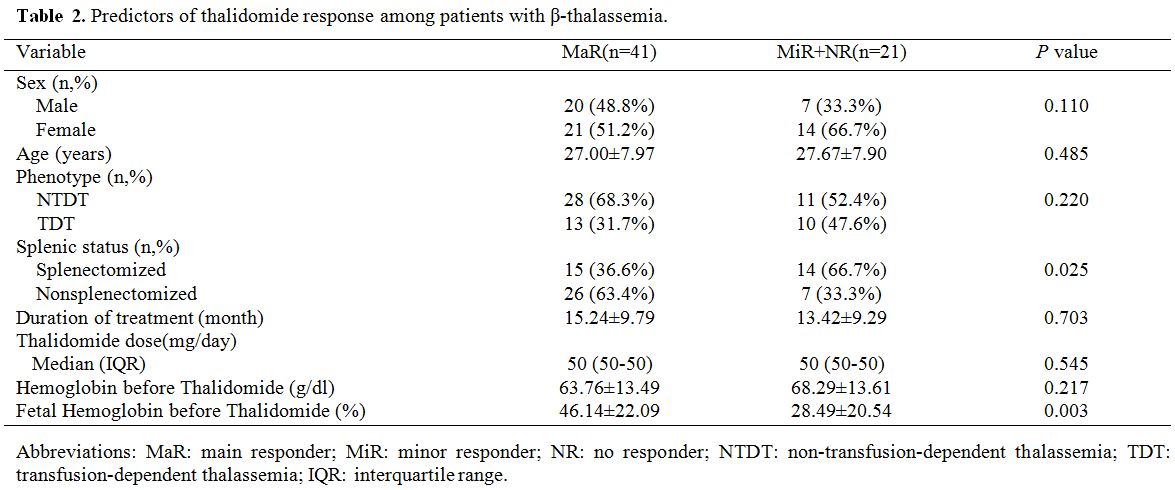 |
Table 2. Predictors of thalidomide response among patients with β-thalassemia. |
Toxicity.
The mild adverse effects of thalidomide were recorded in 10 patients.
The most common toxicity was at the gastrointestinal level (5/62)
followed by a rash (2/62) and menstruation disorders (2/62), but the
symptoms were transient and recovered after symptomatic treatment or
temporary drug discontinuance. During long-term follow-up, one patient
developed peripheral neurotoxicity with intermittent numbness of both
lower limbs. Potential underlying conditions or diseases responsible
for peripheral neurotoxicity were ruled out. The initial dose of
thalidomide administered was 100 mg/d for 23 months, and the cumulative
dose was ~ 55 g. Distal numbness of both lower limbs occurred about 18
months after therapy, and these symptoms were incompletely reversed
after drug withdrawal over a subsequent 4-month period. During the
treatment period, no hematological toxicity or bone marrow suppression
was detected in patients. Furthermore, thalidomide had no unfavorable
effects on liver or kidney function and induced no significant changes
in alanine aminotransferase (ALT) or creatinine (Cr) levels.
Discussion
In
this study, we analyzed the efficacy and safety of thalidomide for
patients with β-thalassemia using a relatively large cohort and
long-term follow-up, with encouraging results. Primary data showed
significant efficacy of thalidomide for β-thalassemia, with the rate of
response for thalidomide being much better than that reported for
hydroxyurea treatment.[22] Guangxi Zhuang Autonomous
Region, Southern China, is an area with a high prevalence of
thalassemia and is economically underdeveloped.[23] Many patients with thalassemia are not sufficiently transfused due to a shortage of blood products.[24]
Moreover, compared to transfusions, thalidomide is more convenient and
economically more feasible in China. Actually, transfusions induce iron
overload, which can be prevented by expensive drug chelation,[25] mostly in the heart but only partially in endocrine organs.[26]
In addition, we observed a long-lasting effect of thalidomide for
β-thalassemia in long-term follow-up relative to earlier short-term
case reports.[15,16] Compared with the decline in hematological response after hydroxyurea treatment for 12 months,[9,10]
the efficacy of thalidomide was stable over time, and no reduction of
the hematological reaction was observed during long-term follow-up,
which would be of considerable benefit to patients requiring continued
treatment. Unfortunately, we have not observed a decline in SF levels
after treatment.
Although the use of thalidomide to treat
β-thalassemia has achieved good results, there is still no consensus on
the optimal and maintenance dose for clinical application. At present,
the therapeutic dose of thalidomide for β-thalassemia is ~50-100 mg/d,[15-17,19,20]
and the dose-response relationship is yet to be established. Compared
with a daily dose of 50 mg/d, we observed that the dose increment did
not give significant added benefit. For responders who received
maintenance therapy at a relatively low dose, the response was evident
despite a decrease in Hb levels. Interestingly, Hb decreased rapidly to
the baseline level after drug withdrawal and was restored after the
re-introduction of the drug, consistent with the results of Fozza at
el.[15] Therefore, thalidomide seems to have a
“switching effect” on β-thalassemia, and the maintenance dose could be
reduced in the future. Since patients with thalassemia require lifelong
medication, it may be valuable to compare the effects of thalidomide
with a different maintenance dose.
Limited complications were
reported in β-thalassemia, with peripheral neuropathy, which is one of
the most feared complications, being documented in one patient. The
mechanism of thalidomide-induced neurotoxicity remains to be clarified.
Several studies have explored risk factors for thalidomide neuropathy,
including age, daily dose, duration of drug exposure, cumulative dose,
and preexisting neuropathy.[27] The incidences of
peripheral neurotoxicity that have been reported are relatively low and
are limited at a daily dose of >50 mg.[27-29]
Although only one patient presented with peripheral neurotoxicity in
our study, physicians contemplating its use should be vigilant to its
occurrence and take preventive action. Furthermore, it is valuable to
explore lower therapeutic or maintenance doses to maximize the
benefit/risk rate.
The current study is the first to assess the
efficacy and safety of thalidomide in a relatively large cohort over a
long-term follow-up period and provide new insights on the continued
efficacy and safety of thalidomide in patients with β-thalassemia.
A
limitation of this study was its design, having included patients from
three centers. Nevertheless, the effects of this limitation were
attenuated by the fact that all centers used 1) the same inclusion
criteria, 2) equal treatment and management of patients, and 3) the
long duration of follow-up.
Conclusions
Thalidomide
is a promising modality of treatment in patients with β-thalassemia. It
can significantly improve Hb levels minimizing the need for blood
transfusion. However, several issues remain to be resolved, such as
establishing the optimal maintenance dose to further improving the
curative effect while avoiding long-term complications.
Acknowledgments
We are grateful to our patients for participating in this study.
Author contribution
All
authors examined the available material, wrote the review, reviewed and
revised the manuscript and provided their approval of the final version
of the manuscript. All authors agree to be accountable for all aspects
of the work.
References
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018. 391(10116): 155-167. https://doi.org/10.1016/S0140-6736(17)31822-6
- Camaschella C, Cappellini MD. Thalassemia intermedia. Haematologica. 1995. 80(1): 58-68.
- Taher
AT, Radwan A, Viprakasit V. When to consider transfusion therapy for
patients with non-transfusion-dependent thalassaemia. Vox Sang. 2015.
108(1): 1-10. https://doi.org/10.1111/vox.12201 PMid:25286743 PMCid:PMC4302976
- Musallam
KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with
fetal hemoglobin induction therapy in patients with β-thalassemia.
Blood. 2013. 121(12): 2199-212; quiz 2372. https://doi.org/10.1182/blood-2012-10-408021 PMid:23315167
- Wu
Y, Zeng J, Roscoe BP, et al. Highly efficient therapeutic gene editing
of human hematopoietic stem cells. Nat Med. 2019. 25(5): 776-783. https://doi.org/10.1038/s41591-019-0401-y PMid:30911135 PMCid:PMC6512986
- Ch
A, Alimirzoyeva Z, Hasanova M, Mammadova T, Shirinova A. Clinical
application of recombinant erythropoietin in beta-thalassaemia
intermedia. Georgian Med News. 2016. (255): 86-92.
- El-Beshlawy
A, El-Ghamrawy M, EL-Ela MA, et al. Response to hydroxycarbamide in
pediatric β-thalassemia intermedia: 8 years' follow-up in Egypt. Ann
Hematol. 2014. 93(12): 2045-50. https://doi.org/10.1007/s00277-014-2154-5 PMid:25062719
- Karimi
M, Haghpanah S, Farhadi A, Yavarian M. Genotype-phenotype relationship
of patients with β-thalassemia taking hydroxyurea: a 13-year experience
in Iran. Int J Hematol. 2012. 95(1): 51-6. https://doi.org/10.1007/s12185-011-0985-6 PMid:22180324
- Rigano
P, Pecoraro A, Calzolari R, et al. Desensitization to hydroxycarbamide
following long-term treatment of thalassaemia intermedia as observed in
vivo and in primary erythroid cultures from treated patients. Br J
Haematol. 2010. 151(5): 509-15. https://doi.org/10.1111/j.1365-2141.2010.08397.x PMid:20955403
- Mancuso
A, Maggio A, Renda D, Di Marzo R, Rigano P. Treatment with
hydroxycarbamide for intermedia thalassaemia: decrease of efficacy in
some patients during long-term follow up. Br J Haematol. 2006. 133(1):
105-6. https://doi.org/10.1111/j.1365-2141.2006.06002.x PMid:16512837
- Ehsani
MA, Hedayati-Asl AA, Bagheri A, Zeinali S, Rashidi A.
Hydroxyurea-induced hematological response in transfusion-independent
beta-thalassemia intermedia: case series and review of literature.
Pediatr Hematol Oncol. 2009. 26(8): 560-5. https://doi.org/10.3109/08880010903271671 PMid:19954365
- Stewart AK. Medicine. How thalidomide works against cancer. Science. 2014. 343(6168): 256-7. https://doi.org/10.1126/science.1249543 PMid:24436409 PMCid:PMC4084783
- Jalali
FMA, Dehghani FA, Hajizamani S, Mossahebi-Mohammadi M, Yaghooti H, Saki
N. Thalidomide is more efficient than sodium butyrate in enhancing
GATA-1 and EKLF gene expression in erythroid progenitors derived from
HSCs with β-globin gene mutation. Int J Hematol Oncol Stem Cell Res.
2016. 10(1): 37-41.
- Aerbajinai W, Zhu J,
Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin gene
expression through increased reactive oxygen species-mediated p38 MAPK
signaling and histone H4 acetylation in adult erythropoiesis. Blood.
2007. 110(8): 2864-71. https://doi.org/10.1182/blood-2007-01-065201 PMid:17620452 PMCid:PMC2018668
- Fozza
C, Pardini S, Giannico DB, et al. Dramatic erythroid response to
low-dose thalidomide in two patients with transfusion independent
thalassemia and severe post-transfusional alloimmune hemolysis. Am J
Hematol. 2015. 90(7): E141. https://doi.org/10.1002/ajh.24030 PMid:25850682
- Ricchi
P, Costantini S, Spasiano A, et al. The long-term and extensive
efficacy of low dose thalidomide in a case of an untransfusable patient
with Non-Transfusion-Dependent Thalassemia. Blood Cells Mol Dis. 2016.
57: 97-9. https://doi.org/10.1016/j.bcmd.2016.01.003 PMid:26810455
- Chen
J, Zhu W, Cai N, Bu S, Li J, Huang L. Thalidomide induces haematologic
responses in patients with β-thalassaemia. Eur J Haematol. 2017. 99(5):
437-441. https://doi.org/10.1111/ejh.12955 PMid:28850716
- Li
Y, Ren Q, Zhou Y, Li P, Lin W, Yin X. Thalidomide has a significant
effect in patients with thalassemia intermedia. Hematology. 2018.
23(1): 50-54. https://doi.org/10.1080/10245332.2017.1354427 PMid:28718348
- Aguilar-Lopez
LB, Delgado-Lamas JL, Rubio-Jurado B, Perea FJ, Ibarra B. Thalidomide
therapy in a patient with thalassemia major. Blood Cells Mol Dis. 2008.
41(1): 136-7. https://doi.org/10.1016/j.bcmd.2008.03.001 PMid:18439858
- Masera
N, Tavecchia L, Capra M, et al. Optimal response to thalidomide in a
patient with thalassaemia major resistant to conventional therapy.
Blood Transfus. 2010. 8(1): 63-5. https://doi.org/10.7175/cmi.v4i2.531
- Ren
Q, Zhou YL, Wang L, et al. Clinical trial on the effects of thalidomide
on hemoglobin synthesis in patients with moderate thalassemia
intermedia. Ann Hematol. 2018. 97(10): 1933-1939. https://doi.org/10.1007/s00277-018-3395-5 PMid:29931453
- Kosaryan
M, Zafari M, Alipur A, Hedayatizadeh-Omran A. The effect and side
effect of hydroxyurea therapy on patients with β-thalassemia: a
systematic review to December 2012. Hemoglobin. 2014. 38(4): 262-71. https://doi.org/10.3109/03630269.2014.927770 PMid:25023087
- Xiong
F, Sun M, Zhang X, et al. Molecular epidemiological survey of
haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern
China. Clin Genet. 2010. 78(2): 139-48. https://doi.org/10.1111/j.1399-0004.2010.01430.x PMid:20412082
- Yin
XL, Wu ZK, He YY, Zhou TH, Zhou YL, Zhang XH. Treatment and
complications of thalassemia major in Guangxi, Southern China. Pediatr
Blood Cancer. 2011. 57(7): 1174-8. https://doi.org/10.1002/pbc.23101 PMid:21394896
- Jialian
L, Yong L, Xue L, Jiayou Z. Economic Evaluation of Chelation Regimens
for β-Thalassemia Major: a Systematic Review. Mediterr J Hematol Infect
Dis. 2019. 11(1). https://doi.org/10.4084/mjhid.2019.036 PMid:31308912 PMCid:PMC6613630
- De
Sanctis V, Soliman AT, Canatan D, et al. Thyroid Disorders in
Homozygous β-Thalassemia: Current Knowledge, Emerging Issues and Open
Problems. Mediterr J Hematol Infect Dis. 2019. 11(1): e2019029. https://doi.org/10.4084/mjhid.2019.029 PMid:31205633 PMCid:PMC6548211
- Baas
P, Boogerd W, Dalesio O, Haringhuizen A, Custers F, van Zandwijk N.
Thalidomide in patients with malignant pleural mesothelioma. Lung
Cancer. 2005. 48(2): 291-6. https://doi.org/10.1016/j.lungcan.2004.10.005 PMid:15829331
- Bastuji-Garin
S, Ochonisky S, Bouche P, et al. Incidence and risk factors for
thalidomide neuropathy: a prospective study of 135 dermatologic
patients. J Invest Dermatol. 2002. 119(5): 1020-6. https://doi.org/10.1046/j.1523-1747.2002.19502.x PMid:12445187
- Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat Rev. 2014. 40(7): 872-82. https://doi.org/10.1016/j.ctrv.2014.04.004 PMid:24830939
[TOP]