Maria Gabriella Mazzucconi1, Erminia Baldacci2, Antonietta Ferretti3 and Cristina Santoro2.
1 Ematologia, Università Sapienza, Roma, Italia.
2 Ematologia, Azienda Ospedaliera Universitaria Policlinico Umberto I, Roma, Italia.
3 Ematologia, Dipartimento Medicina Traslazionale e di Precisione, Università Sapienza Roma, Italia.
Correspondence to: Professor Maria Gabriella Mazzucconi.
Ematologia. Sapienza Università di Roma.Via Benevento 6, 00161
Roma-Italy. Tel: +39 06 857951, Mobile +39 3391773714. E-mail:
mazzucconi@bce.uniroma1.it ORCID ID: 0000-0002-7027-2867
Published: July 1, 2020
Received: May 11, 2020
Accepted: June 18, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020045 DOI
10.4084/MJHID.2020.045
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Acquired
Haemophilia A is a rare acquired bleeding disorder caused by Factor
VIII autoantibodies, which neutralise FVIII activity. These inhibitors
differ from alloantibodies against FVIII, which can occur in congenital
Haemophilia A after repeated exposures to plasma-derived or recombinant
FVIII products. In most cases, the disease occurs suddenly in subjects
without a personal or familiar history of bleedings, with symptoms that
may be mild, moderate, or severe. However, only laboratory alterations
are present in ̴ 30% of patients. The incidence varies from
1 to 4 cases per million/year; more than 80% of patients are elderly,
males and females are similarly affected. There is a small peak of
incidence related to pregnancy in young women aged 20–40 years. The
disease may be underdiagnosed in the elderly. The diagnostic algorithm
is based on an isolated prolonged activated partial thromboplastin
time, normal thrombin time, absence of Lupus Anticoagulant, and a
mixing test that reveals the presence of an inhibitor: the finding of
reduced FVIII activity and the detection of neutralising autoantibodies
against FVIII lead to the diagnosis. The disease is idiopathic
in 44%-63% of cases, while in the other etiological factors
are present. Bleeding prevention and treatment are based on therapeutic
tools as by-passing agents, recombinant porcine FVIII concentrate or,
in a limited number of cases, FVIII concentrates and desmopressin. As
soon as the diagnosis has been made, immunosuppressive therapy must be
started to eradicate the inhibitor. Better knowledge of the disease,
optimal management of bleeding and eradication of the inhibitor have
significantly reduced morbidity and mortality in most patients.
|
Introduction
Acquired
haemophilia A (AHA) is a rare acquired bleeding disorder due to
the development of autoantibodies directed against different epitopes
of Factor VIII (FVIII) molecule, so causing the neutralisation of the
FVIII coagulant activity (FVIII:C), and thus miming a situation similar
to that of congenital haemophilia A (HA). Neutralising inhibitors of
AHA differ from alloantibodies against FVIII of HA patients: in fact,
alloantibodies occur after repeated exposures to plasma-derived or
recombinant FVIII products administered as replacement therapy. The
cause of AHA is due to a breakdown of the immune control mechanism
(immune-tolerance) for both genetic and environmental factors.[1-4]
Generally, autoantibodies are immunoglobulins G (IgG). In most cases,
the disease occurs suddenly in subjects without a personal or familiar
history of spontaneous bleedings and manifests itself with haemorrhagic
symptoms that may be mild, moderate or severe. However, in about 30% of
cases at the beginning of the disease, only laboratory alterations
occur, namely an isolated prolonged activated partial thromboplastin
time (aPTT). In many cases, AHA patients are admitted to the emergency
or general medicine departments, where physicians are not specialists
in the field. For this reason, the diagnosis may be delayed, and the
patients may receive suboptimal treatment. On the contrary, immediate
consultation with a Haemophilia Centre with expertise in the management
of inhibitors against coagulation factors should be required,
regardless of clinical features at presentation.[5,6]
Incidence-Epidemiology
The
incidence of AHA increases with age: it is sporadic in childhood, rare
in adults, but more common in people older than 65. According to the
most extensive available case series, the median age at presentation
was 74[7] and 78,[8] respectively:
more than 80% of patients were 65 years or older. The average incidence
per million/year has been reported to be 0.3 in subjects aged 16-64, 9
in those aged 65-84 and 14.7 in those aged 85 or more.[9] The incidence in children under 16 years old is estimated to be 0.045 million/year.[8]
The age distribution is typically biphasic with a small peak between 20
and 30 years and a larger pick between 68 and 80 years and over. Males
and females are similarly affected, but in the extensive European
Acquired Haemophilia Registry (EACH2) cohort, comprising 501 patients,
a slight males' prevalence was found, that is 53.1% versus 46.9%,
resulting in a male/female ratio of 1:0.88.[7] There
is also a small peak related to pregnancy in young women aged 20–40
years: incidence of AHA in pregnancy within the United Kingdom was
reported to be 1 case/350 000 births.[1,8] In summary, the literature reports an overall incidence of AHA from 1 to 4 cases per million/year[10] or 1-1.5 per million/year;[11] AHA is likely to be underdiagnosed in the elderly.
Diagnosis
Diagnosis
should be considered on the basis of a prolonged isolated aPTT, not
corrected by incubating equal volumes of patient and normal plasma
(mixing test),[12] normal prothrombin time (PT) and
absence of Lupus Anticoagulant (LA). The diagnosis is confirmed by the
finding of reduced FVIII:C levels and by the detection of neutralising
autoantibodies directed against FVIII:C utilising the Bethesda
Nijmegen-modified assay, which allows the titration of the autoantibody
in Bethesda Units/mL (BU/mL).[13,14] Inhibitors are
time-and temperature-dependent, so in some cases, inhibition cannot be
immediately demonstrated by the mixing test, but after a two-hour
incubation at 37°C. In most cases, AHA autoantibodies are type 2
inhibitors and exhibit complex kinetics of inhibition and do not
neutralise FVIII:C entirely, while alloantibodies of HA are generally
of type 1 inhibitor and have second-order kinetics and inactivate
FVIII:C completely. Heat treatment of the sample before assay
(58°C for 90 minutes) may improve inhibitor detection sensitivity by
eliminating residual FVIII:C; an anti-FVIII enzyme-linked immunosorbent
assay (ELISA), particularly after thermal treatment of the samples, has
also been proposed.[15,16] It is crucial to consider
the presence of lupus anticoagulant (LA), which sometimes coexists with
FVIII inhibitor: dilute Russel viper venom time (dRVVT) test is useful
for LA detection in these cases. Heparins, heparinoids, and direct oral
anticoagulants may interfere with inhibitor test results and mimic
circulating inhibitors. Summarising, AHA is defined by the presence of
a neutralising FVIII inhibitor >0.6 BU/mL and a FVIII:C <50%[17,18] (Figure 1).
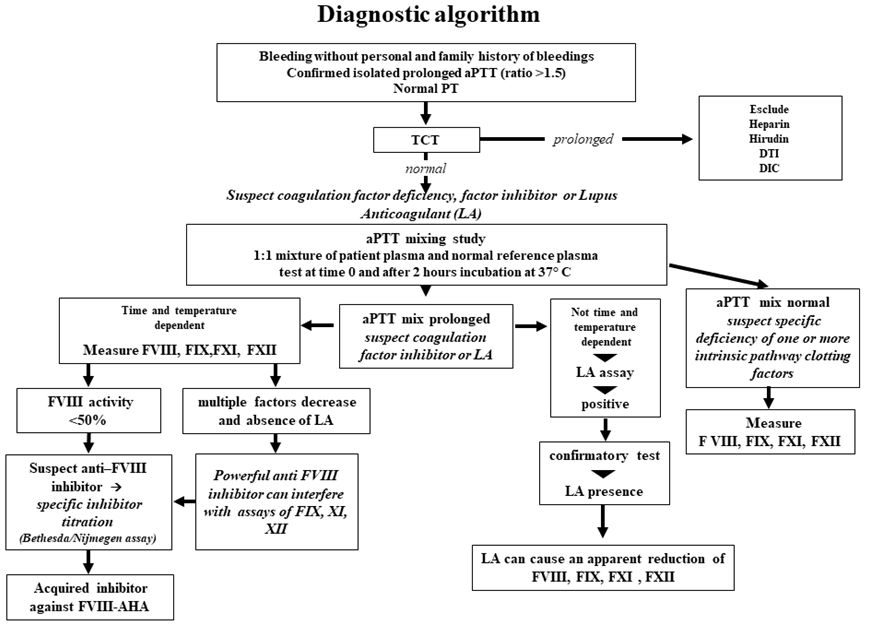 |
Figure 1. |
Pathogenesis
Inhibitors against FVIII can be identified in about 20% of healthy donors:[19]
these are directed against FVIII:C in pooled normal plasma, but not in
autologous plasma, so, they are alloantibodies targeting an
unidentified polymorphism and lacking clinical significance.[20] Autoantibodies in AHA occur for a breakdown of immune tolerance mechanisms, that regulate normal immune response to FVIII[21]
and represent a polyclonal IgG population. A complex interaction
between different CD4+ T cell subsets is implicated in the production
of anti-FVIII antibodies: Th1cells which stimulate B cells to produce
IgG1 antibodies and Th2 cells which stimulate B cells to produce IgG4
antibodies. Moreover, a correlation between the proportion of IgG4
anti-FVIII antibodies and high inhibitor titre has been found.[22,23]
Autoantibodies of AHA are similar to alloantibodies of HA and IgG4 is
usually a major component of the antibody population, although IgG4
represents only 5% of the total IgG in normal plasma; IgG1 and IgG2,
and less often IgG3, are also present.[20] Isotypes
and IgG subclasses were evaluated in a large AHA population at
baseline: IgG4-subclass autoantibodies were found in 98% of cases, IgG1
in 88%, IgG2 in 77% and IgG3 in 41%; IgA and IgM autoantibodies
were detected in 46% and 9% of cases, respectively. IgG4 and IgG1
correlated with the highest inhibitor titre. IgA autoantibodies that do
not neutralise FVIII:C are potential predictors of recurrence and poor
outcome of AHA, while IgG subclasses do not.[24]
Inhibitor antibodies in AHA and HA are mainly directed against the A2
and C2 epitopes of FVIII molecule, but in AHA either anti-A2 or anti-C2
domain, autoantibodies are recognized, not both. The C2 domain is more
frequently targeted.[20] Anti–FVIII C1 domain
antibodies in AHA and HA were recently studied and were found in 78% of
AHA and 57% of HA patients, respectively, but their clinical relevance
is unclear.[25] It seems that global coagulation is
more suppressed in AHA than in severe HA due to the inhibition of
Factor IX activated (FIXa)-dependent Factor X(FX) activation in the
presence of anti-C2 autoantibodies against FVIII.[26] A study reported a stronger statistically significant response of autoantibodies against the A1a1-A2a2-B fragments of FVIII, particularly against the A1a1
domain, in the post-partum AHA group compared with the other AHA
patients' groups. IgG4 subclass was predominant in all groups, but the
anti-A1a1-A2a2-B and the anti-A1a1domains
autoantibodies of the IgG1 and IgG3 subclasses were more frequently
detected in post-partum AHA than in the other AHA groups. This finding
may be due to the post-partum greater involvement of the Th1-driven
response, while in the other AHA groups generation of Th2-driven IgG4
seemed to be predominant. This kind of Th1-driven response may
contribute to a successful outcome of post-partum AHA.[27]
Bleeding Pattern
Clinical
features of AHA differ from those of HA because bruising,
retroperitoneal, muscle, gastrointestinal and urogenital bleedings are
frequent, whereas haemarthroses are uncommon. Compartment syndrome and
compression of nerves and blood vessels may also be found.[28] Gastrointestinal, intracranial and retroperitoneal haemorrhages are often fatal.[8]
In most cases, bleedings occur suddenly, while in about 25% of the
cases they are caused by a trauma or an invasive procedure.[29]
Sometimes, AHA has been found in subjects receiving anticoagulant or
antiplatelet drugs: in these cases, the diagnosis may be delayed,
because the bleeding is assumed to be caused by these agents.[28]
Therefore, excessive bruising or unexpected bleeding in patients taking
antiplatelet or anticoagulant medications should be further
investigated, especially in older adults, and whether evidence of an
overdose of such drugs is lacking based on laboratory tests. In
the past mortality related to bleeding had been reported to be 22-31%.[30,31] However, an incidence between 3% and 9% was found more recently, in particular, 3.2% in the EACH2 cohort,[7,8,32] maybe due to improved therapeutic approach, but mortality caused by infections appears to be increased.[33]
Associated Diseases/Conditions
In
about 50% of cases, there are no underlying diseases or conditions
associated with AHA; thus it has been described as idiopathic in many
large case series at a rate ranging from 43.6% to 63.3%:[1,7,8,30]
the EACH2 study reported that the disease was idiopathic in 51.9% of
501 patients. In comparison, in all other cases, etiological factors
were autoimmune diseases (11.6%), malignancy (11.8%), pregnancy (8.4%),
infections (3.8%), use of drugs (3.4%), monoclonal gammopathy of
undetermined significance (2.6%), rheumatic polymyalgia (2.2%),
dermatological diseases (1.4%), blood products transfusion (0.8%), and
other disorders (2%).[7] The most common solid cancers are prostate cancer, followed by lung cancer.[34,35] Pregnancy-related AHA occurs mainly in the post-partum period, between 3 and 150 days after delivery[36] mostly after the first pregnancy, but the inhibitor is sometimes recognised during pregnancy in 2.5–14% of patients.[36,37]
Moreover, it may become evident during labour, causing severe,
unexpected bleeding; the transplacental passage of the
autoantibodies to the foetus can lead to foetal bleeding.[22,38] Considering the largest available AHA cohorts, pregnancy-related AHA ranges between 2% and 15%.[8,30,39,40] In most cases, the inhibitor titre is rather low (median about 20 BU/mL).[37,41]
The prognosis is favourable, with a low mortality rate (0-6%) and
a high percentage of remission (77-86%), in some cases, even
without treatment.[1,29,40]
.
Case Series
We considered eight case series published from 1981 to 2018, each including 40 or more evaluable patients with AHA.[1,7,8,30,42,43,44,45] One thousand four hundred eleven patients were recruited. Males' prevalence was found in 5 studies, ranging from 58% to 68%,[7,42-45] the prevalence was in favour of females in 2 studies, 55% and 57%, respectively,[1,8] while in the other one almost equal number of males and females was reported.[30] In 7 studies median patients' age ranged from 64 to 78 years[1,7,8,42-45] and in the other one, most patients were over 50 years;[30] in the French study, 69% of patients were over 70 years.[42]
The incidence of AHA becomes more frequent with increasing age, but the
likelihood of finding an underlying disease seems to decrease with age.[8] No AHA-associated disease was found in a median of 52% (46%-67%) of patients,[7,8,30,42-45] while the most frequent underlying disorders were autoimmune diseases and cancer, in young and older people, respectively.[42] Pregnancy-related AHA was reported in 7/8 case series, with a rate ranging from 2% to 15%.[1,7,8,30,42-44] Excluding pregnancy-related cases, AHA was described in young or very young people in 3 studies only.[1,8,30]
Severe bleedings at diagnosis were found to be 60%, 70% and 87% in 3 studies, respectively.[7,30,45] Mortality rate bleeding-related ranged from 2.9% to 9% (median 4.0%),[7,8,42,43] while mortality rate disease-related was reported to be 22%[30] and 11%,[1]
respectively. Some prognostic factors were also identified. Age >65
years vs < 65, related diseases (malignancy vs post-partum vs
others), inhibitor titre at diagnosis (>10 BU/mL vs <10 BU/mL),
the achievement of inhibitor eradication (no vs yes) had a significant
negative impact on overall survival (OS) on univariate analysis, but
only inhibitor eradication, age at diagnosis and underlying diseases
had a consistent, independent significant prognostic value on
multivariate analysis. Regarding disease-related survival, the same
four variables showed a significant prognostic value on univariate
analysis, but only inhibitor eradication and age remained statistically
significant on multivariate analysis.[1] Age appeared to be the only prognostic factor associated with survival in the UK study.[8]
Age over the median of the studied group (76.3 years), low haemoglobin
level at diagnosis, presence of neoplasia and failure of inhibitor
eradication were significant negative prognostic factors in the EACH2
study, while gender, inhibitor titre and FVIII:C did not.[7]
A study, aimed at identifying risk factors in patients treated with a
uniform immunosuppressive regimen for inhibitor eradication, showed
that the time to partial response to therapy did not depend
significantly on age, gender, underlying disorder, and poor performance
status (PS), i.e. WHO-PS
>2; baseline FVIII:C<1% was significantly associated with time to
partial response, while inhibitor titre >20 BU/mL did not; only
baseline FVIII:C <1% remained significantly related with time to
partial response on multivariate analysis. Baseline FVIII:C<1% and
WHO-PS>2 were significantly associated with a lower rate of complete
response to therapy, both on univariate and multivariate analysis.
Patients with poor PS were more likely to die before achieving a
complete response. Baseline FVIII:C <1%, inhibitor >20 BU/mL, age
>74 years, WHO-PS >2, male gender, malignancy and renal failure
were associated with a poor OS on univariate analysis, but only three
baseline factors remained independent predictors of poor OS on
multivariate analysis: FVIII:C <1%, WHO-PS >2, and malignancy.[43]
In the same patients' population presence of anti-FVII, I IgA
autoantibodies were identified as a potential predictor of recurrence
and poor outcome of AHA.[24]
Specific AHA Populations
Some
publications are mainly addressed to specific AHA populations:
children, older people and pregnant or post-partum women. As for
children, a review[38] showed that 42 cases of inhibitors against coagulation factors were collected in childhood: 37 reported de novo
inhibitors and five transplacental transmissions of maternal
inhibitors; the M/F ratio was 1.1. The inhibitor was directed to FVIII
in 28/37 cases (75.7%). An underlying autoimmune disorder was found in
16.7% of cases, but the inhibitor was frequently associated with
infections (16.7%) or use of antibiotics (22.2%), especially penicillin
or penicillin-like antibiotics; 33.3% of cases were idiopathic. The
outcome was favourable: the inhibitor disappeared in 80.6% of cases,
after a median of 2.5 months, the highest remission rate (100%) was
associated with infections or antibiotics use. Other cases have been
described regarding 7 children (4 males, 3 females) diagnosed with AHA
at a median age of 10 years (range 5-14).[46-52] Symptoms described at diagnosis were muscle haematomas,[46,47,52] ankle haemarthrosis,[46] severe bleeding after tooth extraction.[49] Associated conditions were: previous HSCT and concomitant staphylococcus aureus infection,[50] streptococcal infection and amoxicillin treatment,[51] previous course of amoxicillin.[52]Two papers have recently regarded AHA in the elderly: one review from 80 studies, including 159 cases[53] and a cases report concerning a small number of patients.[54]
In the first one, most patients were men (64%) with a mean age of 76.1.
Mortality was high, despite the number of therapies used for inhibitor
eradication, probably due to the lack of rapid diagnosis and to
inadequate management and monitoring. The other one described only 6
patients, but, interestingly, 4 were 90 or older: The Authors
underlined that AHA shows a wide variety of symptoms in the elderly,
indicating the need of individualised management. Regarding
pregnancy-related AHA, a survey carried out in 42 Italian Haemophilia
Centres, identified 20 cases of post-partum AHA among 96 patients
(20.8%), diagnosed during 15 years: 19/20 cases were idiopathic, and in
six the inhibitor was identified because of surgical bleeding. Nine
women did not require haemostatic treatment. The inhibitor was
diagnosed for the occurrence of significant bleeding at a median time
after delivery of 60 days (1–150). Eighteen women received treatment
for inhibitor eradication with an excellent outcome. In two patients
without bleedings, the inhibitor disappeared without therapy. No
relapse was recorded in subsequent pregnancies occurred in 4 women.[39] In the EACH2 registry,[40]
42 cases (8.4%) of peripartum-associated AHA were diagnosed over 6
years. Evidence of antepartum inhibitors was found in 8 women, and 2
babies had postnatal bleeding, suggesting a transplacental transfer of
the autoantibody. The median time between delivery and diagnosis of AHA
was 89 days (21–120). Bleedings were successfully managed, and most
women achieved inhibitor eradication. In conclusion,
pregnancy-associated AHA is rare, but the awareness of it is crucial
for rapid and appropriate management. Relapses during the subsequent
pregnancies are very rare.[18]
Treatment and Prevention of Bleeding
Patients
with AHA must be treated immediately as soon as major bleeding occurs
or haemoglobin levels decrease significantly. Prophylactic haemostatic
treatment must be given in subjects at high risk of bleeding (surgery,
delivery, peptic ulcer, etc.). Invasive procedures should be avoided if
not strictly necessary. Replacement therapy with plasma-derived or
recombinant FVIII concentrates is not effective in the presence of high
inhibitor titre. In case of low titre (<5 BU/mL), the dose must be
adequately adjusted to overcome the inhibitor. However, the risk of
anamnestic response, that is a rise of inhibitor titre, should be
seriously taken into consideration and careful control of FVIII:C
levels and inhibitor titre is mandatory. Desmopressin (DDAVP), alone or
associated with FVIII concentrates may be useful in case of minor
bleedings if the inhibitor titre is low (<2 BU/mL[18] and basal levels of FVIII:C are over 5%.[22,55,56] In the EACH2 study, FVIII concentrates were successful in controlling bleeding in 69.6% of cases.[57]
Products derived from porcine FVIII were administered in the past, with
good results, because porcine FVIII shows a reduced cross-reactivity
against anti-human FVIII inhibitors, but in 2004 their use was
suspended, for viral safety concerns.[58,59] A
recombinant porcine FVIII (rpFVIII, susoctocog alfa, ObizurR)
concentrate was sub sequentially developed; it has been approved for
the treatment of bleedings in AHA in the United States, Canada and
Europe. It is a glycosylated B-domain deleted, recombinant FVIII with
porcine sequence and low cross-reactivity towards anti-human FVIII
antibodies; it is produced in a well-characterised BHK cell line and
manufactured using two viral clearance steps, solvent detergent and
15-nm nanofiltration.[60,61] It shows a favourable
safety and efficacy profile and therefore constitutes a valid
therapeutic option for the treatment of AHA.[62] In
the first prospective phase 2/3 multicenter, international, open-label
registration study concerning 28 adults with AHA, suffering from
serious bleedings, rpFVIII demonstrated good clinical efficacy,
reaching a bleeding control in 24/28 patients. The response of FVIII:C
levels to rpFVIII infusion depends on the presence of an inhibitor with
cross-reactivity towards porcine FVIII in the patient's plasma.
Patients without cross-reactivity reached very high FVIII activity
levels (118%-522%), after an initial loading dose of 200IU/Kg. For this
reason, it is mandatory to determine baseline concentrations of
anti-porcine FVIII antibodies to predict the effectiveness of rpFVIII.
Moreover, infusion of rpFVIII may trigger an increase of the inhibitor
titre or a de novo occurrence of an anti-rpFVIII inhibitor in some
cases, with a subsequent reduction of efficacy. In this study, 5/28
(17.9%) treated patients, who did not have detectable anti-porcine
FVIII at baseline, developed a de novo anti-rpFVIII inhibitor.
Therefore, accurate search and monitoring of both anti-human and
anti-porcine inhibitors and determination of FVIII:C levels are
mandatory before and during treatment.[63] Two
subsequent publications have shown that lower initial doses of rpFVIII
(100 IU/Kg) achieved the same efficacy as that obtained in the
registration studies with higher doses.[64,65]
However, in the first one, one patient developed a de novo anti-rpFVIII
inhibitor and another suffered from a lower-limbs deep vein thrombosis;
in the second one, a de novo
anti-rpFVIII inhibitor occurred in 2 patients. In both studies, the
Authors highlighted the efficacy of lower doses of rpFVIII and the need
to titrate the rpFVIII doses, using close monitoring of FVIII:C. Before
the availability of the rpFVIII concentrate, the alternative to FVIII
replacement therapy was represented by the products capable of
by-passing the inhibitor activity ("by-passing agents"), namely the
activated prothrombin complex concentrates (APCCs) and the recombinant
factor VII activated (rFVIIa). They circumvent the inadequate
activation of FX. Both rFVIIa and APCCs are effective in the treatment
of bleedings, but no comparative studies are showing greater efficacy
of one product than the other.[22,57]
Many case series have been published concerning the use of both
products. Since1997, treatment with rFVIIa (NovoSeven®) has been
reported either as a first-line therapy tool or after the failure of
other therapeutic approaches.[57,66,67,68,69]
A large number of bleedings were treated with rFVIIa with efficacy
ranging from 83% to 100% and, when it was administered as second-line
therapy, from 66% to 75%.[57,66,67,68,69]
The doses ranged from 40 to 180µg/Kg (average 90) every 2-6 h, for a
variable duration, according to bleeding severity, clinical situation
and disease status.[70] A 10-year post-marketing
surveillance analysis was recently published: NovoSeven® was used in
371 bleeding episodes occurred in 132 AHA patients; efficacy was
recorded in 92% of cases. Interestingly, the response rate was
significantly better in patients who received an initial dose of
≥90µg/Kg than of <90µg/Kg. Treatment with rFVIIa was more effective
if given immediately after the start of bleeding.[69] Regarding APCCs, a retrospective study[71]
described the efficacy of APCC (FEIBA®) in 34 AHA patients: most of
them received FEIBA® at a single dose of 75IU/Kg repeated every 8-12
hours: the complete response was reached in 100% of moderate and in 76%
of severe bleedings, respectively. In the EACH2 study, 19% of bleeds
were treated with APCC and 51% with rFVIIa: both by-passing agents
showed similar efficacy rate (FEIBA® 93%, rFVIIa 91%).[57]
A French registry collected data on 34 AHA patients (mean age 81.8
years) who received FEIBA® for bleeding episodes or prophylaxis for
invasive procedures. Mean initial dose of FEIBA® for acute bleeding was
75.4IU/Kg, mostly administered twice daily. The median duration of
treatment was 4 days. Efficacy was recorded in 88.0% of bleeding
episodes, although 4 patients experienced six serious adverse events
possibly related to FEIBA®.[72] A
retrospective/prospective multicentre Italian study ("FAIR study") was
recently published and regarded 56 patients (median age 69.9 years)
enrolled in 12 Italian Haemophilia Centres. FEIBA® was given as
first-line therapy in 82.2% of cases, at a mean dose of 72.6IU/kg for a
median period of 8 days; efficacy was reached in 96.4% of bleedings.
Antifibrinolytic agents were used with FEIBA® in 39. 6% of treated
cases at clinician's discretion. Low-dose FEIBA® for short-term
prophylaxis (mean dose 54.2IU/Kg), was administered in 26.8% of the
patients after the first episode to prevent bleeding relapse at an
average frequency of 24 hours. In the FAIR study, an anamnestic
response was recorded in 5.9% of cases; no thromboembolic events
occurred.[73] The thromboembolic risk was seriously
considered for both by-passing agents. Sumner et al. reported 12
thrombotic events in 10 patients treated with rFVIIa;[68]
11 thrombotic events (arterial 7 and venous 4) were described in the
EACH2 cohort, with a similar incidence for rFVIIa (2.9%) and APCCs
(4.8%).[57] Amano et al.[69] reported 5 thromboembolic events in 3 elderly patients with comorbidities treated with rFVIIa (2.3%). Tiede et al.[43]
described that the rate of fatal vascular events was 5% in patients
treated only with rFVIIa and 10% in those who received a
combination of rFVIIa and tranexamic acid (TA). Risk of thrombotic
events can increase if both by-passing agents are administered in a
combined or sequential way: thromboses occurred in 5/9 AHA patients
treated with this modality (55%).[74] Interestingly, in French registry[72] and in "FAIR study"[73]
no thromboembolic events were reported. However, careful monitoring of
patients should be performed, especially in the elderly and in those
with comorbidities or predisposing conditions, such as previous
thrombotic events or thrombophilia. Antifibrinolytic drugs, such as TA,
are considered a useful tool for the treatment of bleeding, especially
of mucous origin, except haematuria, in patients with congenital or
acquired bleeding disorders; TA contributes to the clot stability, but
doubts remain about its use in association with APCCs, due to the risk
of thromboembolic complications. Mouthwashes with TA are safe and
effective for mouth bleeds, also during treatment with APCCs. The
association of antifibrinolytics with FEIBA® in AHA patients was
recently evaluated in 39.6% of 101 treated bleedings: no thromboembolic
events were recorded, despite a large portion of patients showed
serious comorbidities or severe cardiovascular diseases. However, these
findings need to be confirmed in proper, larger clinical trials.[75]
In AHA patients with high inhibitor, titre APCCs have been considered a
cost-effective therapy option when compared to rFVIIa.[76]
We can conclude that both by-passing agents are still suitable and
effective first-line treatments of AHA, despite the availability of the
rpFVIII. In rare cases, characterised by serious life-threatening
haemorrhages and a very high inhibitor titre, extracorporeal removal of
the autoantibody by plasmapheresis, or immunoadsorption on
staphylococcal protein A or polyclonal sheep antibodies against human
immunoglobulins has been used to remove the autoantibody temporarily
and allow the administration of FVIIII concentrates.[77]
A novelty in the treatment of HA has been identified in an innovative
drug, emicizumab. Emicizumab is a bi-specific, humanised monoclonal
antibody which bridges FIXa and FX to replace the physiologic function
of missing activated FVIII in HA patients, thereby restoring
haemostasis. Some randomised and non-randomised clinical trials carried
out in adults and in children with HA, with or without inhibitor
against FVIII, have shown remarkable effectiveness of emicizumab,
administered as prophylaxis therapy, in preventing bleedings.[78,79,80,81]
Although the results of these studies have already led to the approval
of treatment with emicizumab in many countries around the world,
including Europe, its use in AHA is not currently allowed. However,
some case reports were recently published describing emicizumab
treatment in some elderly AHA patients. Emicizumab was effective
when given after failure of previous agents, such as rpFVIII and APCC.[82,83,84,85]
Moreover, a recent study has demonstrated, in an experimental model,
that the ex-vivo addition of various concentrations of emicizumab to
plasma samples of AHA patients has been capable of restoring thrombin
generation. Therefore, emicizumab can improve the ex-vivo coagulation
potential in plasma of patients with AHA and, based on these results;
its therapeutic use could also be effective in AHA[86] (Table1).
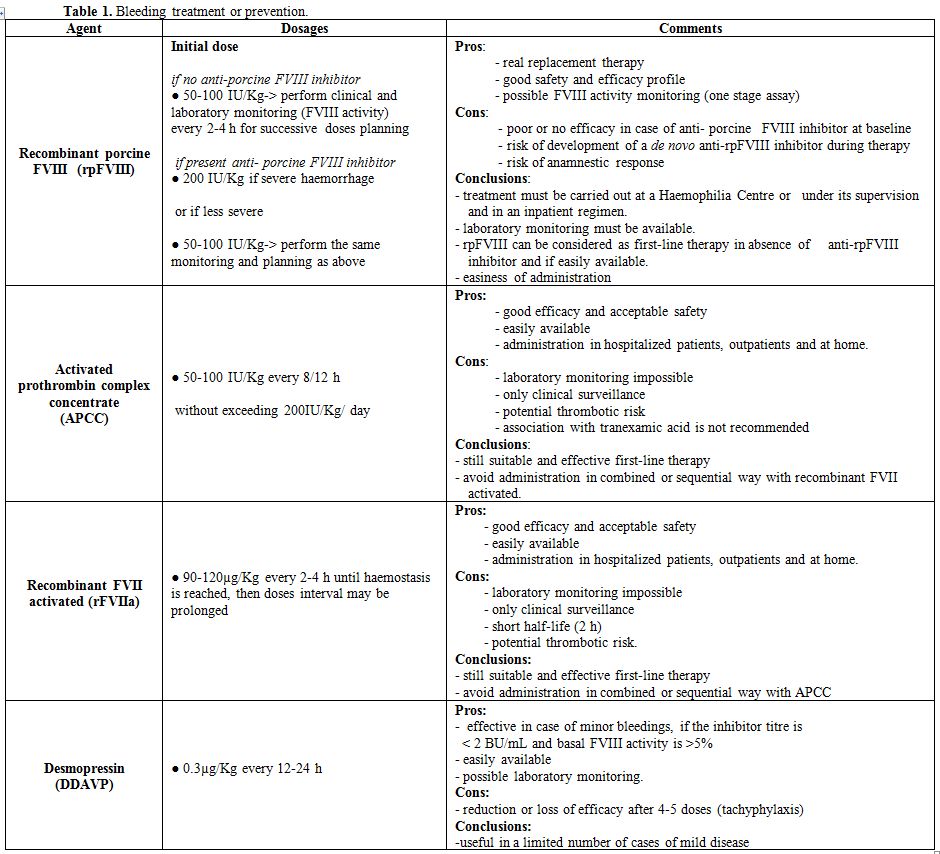 |
Table 1. Bleeding treatment or prevention. |
Inhibitor Eradication: Immunosuppressive Therapy (Ist)
Patients
with AHA, although asymptomatic at diagnosis, remain at risk of
bleeding as long as the inhibitor persists. Although up to 36% of
patients achieve spontaneous disappearance of the autoantibody without
immunosuppressive therapy (IST),[1] the inhibitor must
be eradicated by IST, administered immediately after diagnosis in order
to induce remission of the disease, as soon as possible.Corticosteroids
alone or associated with cyclophosphamide have been used as first-line
treatment, while other immunosuppressants or the monoclonal anti-CD20
antibody, rituximab, have been given as second/third-line therapy. In
the only randomised study published until now, 31 AHA patients were
initially treated with prednisone, if the autoantibody persisted and
there was no rise in FVIII:C, patients were randomised to either
prolong prednisone for other 6 weeks or taper prednisone and start
cyclophosphamide or continue prednisone and add cyclophosphamide. About
1/3 of patients responded to prednisone alone, while in 50% of
prednisone-resistant patients, cyclophosphamide-containing regimens
achieved eradication of the inhibitor. In conclusion, patients should
be initially treated with prednisone, while cyclophosphamide appeared
to be an effective second-line therapy for prednisone-resistant
patients.[87] In a metanalysis concerning 234
patients, cyclophosphamide with or without prednisone was found to be
more effective in inducing inhibitor eradication than corticosteroids
or no immunosuppressive treatment at all. However, the superiority of
cyclophosphamide over prednisone was not confirmed with regard to OS,
probably for increased infection-related mortality due to
haematological toxicity of this drug.[1] In a
prospective, non-randomised study, no statistically significant
difference was found regarding the eradication rate of the inhibitor.
The median time to complete remission (CR), namely FVIII:C normal,
inhibitor undetectable, IST stopped or properly reduced, between
patients treated only with corticosteroids (mostly
prednisolone) and patients receiving corticosteroids combined with
a cytotoxic drug, (mostly cyclophosphamide): CR were 76% and
78%, reached at a median time of 49 and 39 days, respectively.[8]
In another meta-analysis, concerning 359 patients, CR (absence of
FVIII inhibitors and normalisation of FVIII:C) was recorded in
68% of patients treated with prednisone alone, in 82% of those treated
with dual therapy (prednisone and cyclophosphamide or azathioprine) and
in 94% of those treated with combined chemotherapy (prednisone,
cyclophosphamide and vincristine). Inhibitor eradication was more
probably achieved by patients treated with IST than by those untreated
at all; in addition, patients undergoing combination therapy had a
lower risk of death.[88] In the EACH2 study,
first-line IST was evaluated in 294 patients: corticosteroids were
given alone in 142 patients and combined with cyclophosphamide in
83; rituximab-based regimens were administered in
51 patients, 18 patients were treated with other regimens.
Complete remission (CR) was defined as complete disappearance of
inhibitor, factor VIII:C over 70%, IST stopped; stable CR was
considered as persistent CR without relapses during follow-up. The
median time to FVIII:C>70% and undetectable inhibitor in patients
treated with corticosteroids alone were 32 and 34 days, respectively,
while in those receiving corticosteroids and cyclophosphamide 40 days
and 32 days, respectively. Complete remission was reached at a median
time of 108 days in patients receiving corticosteroids alone and of 74
days in those undergoing corticosteroids and cyclophosphamide and CR
rates were 58% and 80% in the two groups, respectively. Complete
remission was obtained by 61% of patients treated with rituximab-based
regimens. Relapses occurred in 18% of patients treated with
corticosteroids alone, while in those receiving combined therapy or
rituximab-based regimens in 12% and 3%, respectively; stable CR was
recorded in 48%, 70% and 59% of each group, respectively. Underlying
disease or sex did not affect the remission; age showed a moderate
influence; on the contrary, baseline low inhibitor level (<16 BU/mL)
and higher FVIII:C were significantly associated with faster inhibitor
eradication and normalisation of FVIII:C level. At last follow-up
(median 262 days), death rates were similar among the groups:
28% in patients treated with corticosteroids only, 33% in those
treated with corticosteroids and cyclophosphamide and 20% in those
treated with rituximab-based regimens; 4 patients receiving
corticosteroids and cyclophosphamide died for sepsis due to the
immunosuppression, one of them was neutropenic.[89]
Another study investigated prospectively standardised IST: 102 patients
received prednisolone initially for 3 weeks; then oral cyclophosphamide
was added if remission was not reached, (weeks 4–6); then rituximab was
given with prednisolone (weeks 7-10) if lack of response. Partial
remission (PR) was defined as FVIII:C>50%, no active bleeding, no
haemostatic drugs for 24 h, CR as PR plus inhibitor absence,
prednisolone tapered to less 15 mg/day and any other immunosuppressive
therapy stopped; PR was achieved by 83% and CR by 61% of patients,
respectively. The median time to PR and CR was 31 days and 79 days,
respectively. Forty-eight % of the patients were alive in stable CR
after a median observation time of 403 days.[43] In
resistant or relapsed patients, other therapeutic approaches have been
experienced: cyclosporine alone or in combination with corticosteroids,
mycophenolate mofetil or multiple immunosuppressive drugs, with
variable results.[18,22] Based on
the experience gained in congenital HA with inhibitor, immunotolerance
induction (ITI) protocols has been proposed in very selected cases for
inhibitor eradication: Budapest protocol[90] and the modified Bonn-Malmo protocol (MBMP).[91]
However, these expensive procedures require ad hoc specialised clinical
departments. High-dose intravenous immunoglobulins (IVIG), alone or in
combination with corticosteroids, are no longer considered suitable for
the inhibitor eradication.[1,8,89]
Rituximab has been used since the early 2000s for the treatment of AHA:
two reviews were recently published on its placement in the first-line
therapy or subsequent lines. Both publications concluded that rituximab
may be considered a safe and useful treatment for AHA, but that it
should be placed on second-line therapy in resistant or relapsed
patients after first-line treatment.[92,93] Rituximab is also effective in pregnancy-related AHA.[94,95,96]Complications
of IST are frequent and sometimes fatal: patients, especially if
elderly, should be monitored for the occurrence of adverse events,
particularly infections. In the UK study, sepsis occurred in 33%
of cases and led to death in 12% of them;[8] in the GTH-AH 01/2010 study, 54 infections were diagnosed in 37/102 patients, and 16/102 died from sepsis;[43] in the SACHA registry death rate for IST was 12%[42] and in the EACH2 study 4.2%.[89]
Complications of corticosteroid therapy include: increased blood sugar
levels (12%), gastroduodenal ulcer (4%), muscle disorders (4%), and
psychiatric disorders (3%).[8,43]
After inhibitor eradication elevated levels of FVIII:C are often
observed and constitute an independent thrombotic risk factor.[97]
In a recent study, a cohort of 111 AHA patients, followed for a median
time of 25.6 months, was evaluated for relapse pattern. In 14% of them,
one or more relapses occurred after remission obtained with IST. Median
time from diagnosis and from the first remission to the first relapse
was 13.4 months and 12.0 months, respectively. Underlying
lymphoproliferative diseases were predictive of relapse; older age and
male gender appeared to be more frequently associated with recurrence,
while FVIII:C and inhibitor levels at diagnosis were not. Moreover,
relapse was not associated with worse OS. The authors suggested that
the patients should be followed up after remission for at least 2 years[98] (Table 2).
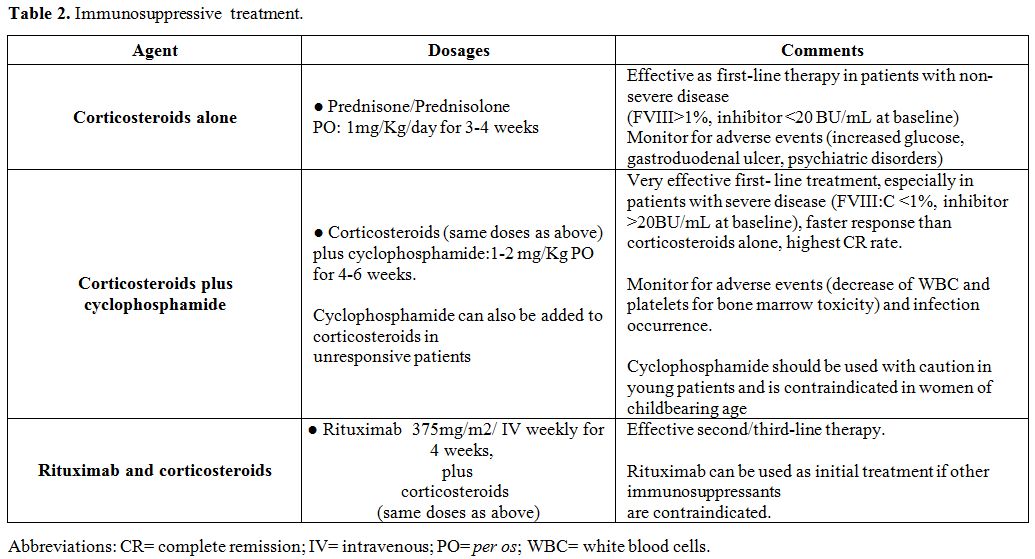 |
Table 2. Immunosuppressive treatment. |
Guidelines
The knowledge of AHA, based on publications of case reports and large case series has led to the development of ad hoc guidelines, as consensus recommendations of experts' panels, regarding diagnosis and therapeutic approach of the disease.[9,18,28,99,100] In 3 of these[28,100,101] the "GRADE system" was used to quote the levels of evidence and the strength of the recommendations.[102]Recommendations and suggestions derived from guidelines are summarised and listed below.Diagnosis: suspect AHA
when a sudden abnormal haemorrhage occurs in subjects, not on
anticoagulation, without personal or family bleeding history, who show
an isolated prolonged aPTT (absence of LA) and a mixing study
consistent with an inhibitor. An unexplained prolongation of aPTT
before surgery or an invasive procedure should always be investigated.
However, in ̴ 30% of cases, only laboratory alterations occur. The diagnosis must be made by a Haemophilia Centre with expertise on coagulation disorders and management of inhibitors against coagulation factors. Test
and monitor anti-FVIII inhibitor with Bethesda Nijmegen-modified assay.
If treatment with rpFVIII concentrate is planned, test and monitor also
anti-prFVIII inhibitors. • Avoid invasive procedures: if necessary, they must be performed in a Haemophilia Centre or after consultation with it.• Look for an underlying cause, or disease as soon as the diagnosis of AHA has been made. Treat any underlying condition.• Treatment of bleeding. Start anti-haemorrhagic therapy in the presence of clinically relevant bleeding symptoms By-passing agents
(APCC, rFVIIa) must be considered as first-line treatment; if the
by-passing agent, administered initially, is ineffective, the other one
should be tried at an early stage. Recombinant and plasma-derived FVIII concentrates and DDAVP
should be reserved to patients with measurable FVIII:C levels and low
inhibitor titre, but accurate laboratory monitoring is necessary; DDAVP is not recommended in the elderly. Porcine recombinant FVIII is also considered as first-line treatment, but its use should be reserved for highly specialised Haemophilia Centres. By-passing agents or rpFVIII should be used in the prevention of bleeding
in the event of surgery or invasive procedure. In exceptional
cases (very severe bleeding and lack of response to standard
treatments), plasmapheresis and/or immunoadsorption, in combination
with high doses of FVIII concentrates can be considered. •
Inhibitor eradication. All AHA patients should receive IST immediately
after diagnosis. First-line treatment should be oral prednisone/prednisolone either alone or associated with oral cyclophosphamide:
this approach allows to reach a CR (persistent undetectable inhibitor,
<0.6 UB/mL, and levels of FVIII:C >70% and IST stopped) in 60-80%
of cases, after a median time of 5-6 weeks. Rituximab can be considered as first-line therapy when standard first-line treatment is contraindicated. Second-line therapies should be attempted if a response to first-line treatment is not reached within 3-5 weeks: rituximab, alone or in combination with corticosteroids, if never given before, cyclosporin, mycophenolate mofetil or multiple immunosuppressive agents. At present, ITI does not appear to be an advisable therapeutic approach. The use of high-dose IVIG is contraindicated. Prognostic markers at baseline (FVIII:C >1% vs < 1% and inhibitor titre >20BU/mL vs < 20BU/mL) should be identified to optimise IST
regarding the combination of corticosteroids with cyclophosphamide or
other immunosuppressants such as rituximab for first-line
therapy. • Monitoring after response to IST: aPTT, FVIII:C and inhibitor titre must be monitored monthly within 6 months, every 2-3 months between 6 and 12 months, and every 6 months after 12 months.• Thromboprophylaxis after response to IST:
mechanical or pharmacological thromboprophylaxis in hospitalised
non-bleeding patients is indicated when FVIII:C is over 50%, while
subjects with prior need for anticoagulation or antiplatelet therapy
can restart it at this moment. Patients showing very high levels of
FVIII:C, during or after IST, should be evaluated for
thromboprophylaxis.• IST in children with AHA:
there are no particular recommendations, given the low frequency of
cases. Anti-haemorrhagic and eradication treatments are similar to
those of adults. • IST in pregnancy-associated AHA: prednisone/prednisolone must be considered as first-line therapy choice; cyclophosphamide and other alkylating agents must be avoided; rituximab is believed to be an appropriate second-line therapy.
Comments and conclusions
Acquired
haemophilia A is a rare and intriguing disease. Its knowledge should be
improved among non-specialised clinicians, because it may suddenly
appear in people otherwise in good health and because the first
approach might occur in emergency departments, where sometimes doctors
do not have experience with the diagnosis and management of this
disease. Ideally, a Haemophilia Centre, with adequate expertise, would
be the best place for the first approach of AHA, but this is not always
possible. Therefore, the establishment of a network would be necessary
on the territory to allow immediate consultation with a reference
Haemophilia Centre, for obtaining both early diagnosis and prompt
therapeutic approach. Over the past years, therapies for bleedings have
gradually improved, thanks to the use of increasingly effective and
more widely available products. The availability of rpFVIII has made
possible a real replacement therapy, thanks to low cross-reactivity of
the rpFVIII with the autoantibody directed against human FVIII.
However, as mentioned above, this treatment should be carefully
monitored and requires to be administered in specialised Centres.
Excellent results can be obtained with IST: first-line therapy with
corticosteroids, alone or combined with cyclophosphamide, has
demonstrated high efficacy; moreover, rituximab in resistant or
relapsed cases or even as first-line approach, when other
immunosuppressants are contraindicated, is currently considered an
effective treatment. In conclusion, the knowledge of the disease has
been improved, therapy for bleeding has reached remarkable results and
IST, set up as soon as possible, offers the possibility of the
inhibitor eradication in most cases. Hence, morbidity and mortality of
AHA have significantly decreased, even in the more advanced age groups.
References
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F,
Villar A. Acquired haemophilia: review and meta-analysis focused on
therapy and prognostic factors. Br J Haematol 2003; 121: 21-35 https://doi.org/10.1046/j.1365-2141.2003.04162.x PMid:12670328
- Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia. 2010;16 (Suppl 3):41-45 https://doi.org/10.1111/j.1365-2516.2010.02259.x PMid:20586801
- Tiede
A, Eisert R, Czwalinna A, Miesbach W, Scharrer I, Ganser A. Acquired
haemophilia caused by non-haemophilic factor VIII gene variants. Ann
Hematol. 2010; 89:607-612. https://doi.org/10.1007/s00277-009-0887-3 PMid:20054547
- Pavlova
A, Zeitler H, Scharrer I, Brackmann HH, Oldenburg J. HLA genotype in
patients with acquired haemophilia A. Haemophilia. 2010; 16:107-112. https://doi.org/10.1111/j.1365-2516.2008.01976.x PMid:20536993
- Collins PW: Treatment of acquired hemophilia A. J Thromb Haemost 2007; 5 (5):893-900. https://doi.org/10.1111/j.1538-7836.2007.02433.x PMid:17461924
- Hay
CR, Brown S, Collins PW, Keeling DM, Liesner R: The diagnosis and
management of factor VIII and IX inhibitors: a guideline from the
United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol
2006; 133 (6):591-605. https://doi.org/10.1111/j.1365-2141.2006.06087.x PMid:16704433
- Knoebl
P, Marco P, Baudo F, Collins P, Huth-Ku¨ hne A, Nemes L, Pellegrini F,
Tengborn L, Le' vesque H, on behalf of the EACH2 Registry Contributors.
Demographic and clinical data in acquired hemophilia A: results from
the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost
2012; 10: 622-31. https://doi.org/10.1111/j.1538-7836.2012.04654.x PMid:22321904
- Collins
PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, Keeling DM,
Liesner R, Brown SA, Hay CR. Acquired hemophilia A in the United
Kingdom: a 2-year national surveillance study by the United Kingdom
Haemophilia Centre Doctors Organisation. Blood 2007; 109: 1870-1877. https://doi.org/10.1182/blood-2006-06-029850 PMid:17047148
- P
Collins, F Baudo, Angela Huth-Kühne, J Ingerslev, C M Kessler, M E
Mingot Castellano, M Shima, J St-Louis and H Lévesque. Consensus
recommendations for the diagnosis and treatment of acquired hemophilia
A. BMC Research Notes 2010; 3:161
http://www.biomedcentral.com/1756-0500/3/161 https://doi.org/10.1186/1756-0500-3-161 PMid:20529258 PMCid:PMC2896368
- M Franchini and G Lippi. Acquired factor VIII inhibitors. Blood; 2008;112 :250-255 https://doi.org/10.1182/blood-2008-03-143586 PMid:18463353
- J
Charlebois, G-É Rivarda, J n St-Louis Management of acquired hemophilia
A: Review of current evidence. Transfusion and Apheresis Science 57;
2018 717-720 https://doi.org/10.1016/j.transci.2018.10.011 PMid:30396835
- Franchini
M, Targher G, Montagnana M, Lippi G. Laboratory, clinical and
therapeutic aspects of acquired haemophilia A. Clin Chim Acta 2008;
395: 14-18 https://doi.org/10.1016/j.cca.2008.05.003 PMid:18505682
- Collins
PW, Percy CL. Advances in the understanding of acquired haemophilia A:
implications for clinical practice. Br J Haematol 2010; 148: 183-194. https://doi.org/10.1111/j.1365-2141.2009.07915.x PMid:19814739
- Verbruggen
B, et al. The Nijmegen modification of the Bethesda assay for factor
VIII:C inhibitors: improved specificity and reliability. Thromb Haemost
1995; 73: 247-251. https://doi.org/10.1055/s-0038-1653759 PMid:7792738
- Sahud
MA, Pratt KP, Zhukov O, Qu K, Thompson AR. ELISA system for detection
of immune responses to FVIII: a study of 246 samples and correlation
with the Bethesda assay. Haemophilia. 2007 May;13 (3):317-22. https://doi.org/10.1111/j.1365-2516.2007.01450.x PMid:17498082
- Batty
P, Moore GW, Platton S, Maloney JC, Palmer B, Bowles L, Pasi KJ,
Rangarajan S, Hart DP. Diagnostic accuracy study of a factor VIII ELISA
for detection of factor VIII antibodies in congenital and acquired
haemophilia A. Thromb Haemost. 2015;114:804-811. https://doi.org/10.1160/TH14-12-1062 PMid:26063073
- Tiede
A, Werwitzke S, Scharf RE. Laboratory diagnosis of acquired hemophilia
A: limitations, consequences, and challenges. Semin Thromb Hemost.
2014;40:803-811 https://doi.org/10.1055/s-0034-1390004 PMid:25299927
- Kruse-Jarres
R, Kempton Christine L, Baudo F,. Collins PW, Knoebl P,. Leissinger CA,
Tiede A,. Kessler CM. Acquired hemophilia A: Updated review of evidence
and treatment guidance. Am J Hematol. 2017;92:695-705. https://doi.org/10.1002/ajh.24777 PMid:28470674
- lgiman
M, Dietrich G, Nydegger UE, Boieldieu D, Sultan Y, Kazatchkine MD.
Natural antibodies to factor VIII (anti-hemophilicfactor) in healthy
individuals. Proc Natl Acad Sci USA 1992; 89: 3795-3799. https://doi.org/10.1073/pnas.89.9.3795 PMid:1570298 PMCid:PMC525577
- Lollar
P. Pathogenic antibodies to coagulation factors. Part one: Factor VIII
and factor IX. J Thromb Haemost 2004; 2; 1082-1095. https://doi.org/10.1111/j.1538-7836.2004.00802.x PMid:15219191
- Reding MT. Immunological aspects of inhibitor development. Haemophilia 2006; 12 (Suppl 6): 30-35. https://doi.org/10.1111/j.1365-2516.2006.01363.x PMid:17123391
- Franchini M, Mannucci PM. Acquired haemophilia A: A 2013 update. Thromb Haemost 2013; 110: 1114-1120. https://doi.org/10.1160/TH13-05-0363 PMid:24008306
- Reding
MT, et al. Distribution of Th1- and Th2-induced anti-factor VIII IgG
subclasses in congenital and acquired haemophilia patients. Thromb
Haemost 2002; 88: 568-575. https://doi.org/10.1055/s-0037-1613257 PMid:12362225
- Tiede
A, Hofbauer CJ, Werwitzke S, Knöbl P, Gottstein S, Scharf RE, Heinz J,
Groß J, Holstein K, Dobbelstein C, Scheiflinger F, Koch A, Reipert BM.
Anti-factor VIII IgA as a potential marker of poor prognosis in
acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood.
2016 May 12;127(19):2289-2297. https://doi.org/10.1182/blood-2015-09-672774 PMid:26912467
- Kahle
J, Orlowski A, Stichel D ,. Healey J F,. Parker ET, Jacquemin M ,
Krause M, Tiede A, Schwabe D , Lollar P, and Konigs C. Frequency and
epitope specificity of anti-factor VIII C1 domain antibodies in
acquired and congenital hemophilia A. Blood. 2017;130(6):808-816. https://doi.org/10.1182/blood-2016-11-751347 PMid:28507083 PMCid:PMC5553573
- Matsumoto
T, Nogami K, Ogiwara K , Midori S. A putative inhibitory mechanism in
the tenase complex responsible for loss of coagulation function in
acquired haemophilia A patients with anti-C2 autoantibodies. Thromb
Haemost 2012; 107: 288-301 https://doi.org/10.1160/TH11-05-0331 PMid:22234708
- Lapalud
P, Ali T, Cayzac C, Mathieu-Dupas E, Levesque H, Pfeiffer C, Balicchi
J, Gruel Y, Borg JY, Schved JF, Granier C, Lavigne-Lissalde G. The IgG
autoimmune response in post-partum acquired hemophilia A targets mainly
the A1a1 domain of FVIII. J Thromb Haemost 2012;10: 1814-1822. https://doi.org/10.1111/j.1538-7836.2012.04850.x PMid:22784315
- Collins
PW, Chalmers E, Hart DP, Liesner R, Rangarajan S, Talks K, Williams M,
Hay CR; UK Haemophilia Centre Doctors. Diagnosis and treatment of
factor VIII and IX inhibitors in congenital haemophilia: (4th edition).
UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013; 162:
758-773
- Baudo F, Mostarda G, de
Cataldo F. Acquired factor Ⅷ and factor Ⅸ inhibitors: survey of the
Italian haemophila centers (AICE). Haematologica 2003; 88 Suppl 12:
S93-99 Available from: URL: https://www.researchgate.net/publication/285180247
- Green D, Lechner K: A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb Haemost 1981, 45(3):200-203. https://doi.org/10.1055/s-0038-1650169 PMid:6792737
- Lottenberg
R, Kentro TB, Kitchens CS. Acquired hemophilia. A natural history study
of 16 patients with factor VIII inhibitors receiving little or no
therapy. Arch Intern Med 1987;147:1077-1081. https://doi.org/10.1001/archinte.1987.00370060073014 PMid:3109341
- Charlebois
J, Rivard GÉ, St-Louis J. Management of acquired hemophilia A: Review
of current evidence. Transfus Apher Sci. 2018 Dec;57(6):717-720. https://doi.org/10.1016/j.transci.2018.10.011 PMid:30396835
- Vautier
M, de Boysson H, Creveuil C, Repesse Y, Borel-Derlon A, Troussard X,
Damaj GL, Bienvenu B, Gautier P, Aouba A.Influence of factor VIII level
and its inhibitor titer on the therapeutic response to corticosteroids
alone in the management of acquired hemophilia: a retrospective
single-center study. Medicine (Baltimore) 2016;95 (November (48):
e5232. https://doi.org/10.1097/MD.0000000000005232 PMid:27902587 PMCid:PMC5134779
- Reeves BN, Key NS. Acquired hemophilia in malignancy. Thromb Res. 2012 Apr;129 Suppl 1:S66-8 https://doi.org/10.1016/S0049-3848(12)70019-1
- Sallah S, Wan JY. Inhibitors against factor VIII in patients with cancer. Analysis of 41 patients. Cancer 2001; 91: 1067-1074 https://doi.org/10.1002/1097-0142(20010315)91:6<1067::AID-CNCR1101>3.0.CO;2-4
- Michiels
JJ. Acquired haemophilia A in women post-partum: clinical
manifestations, diagnosis and treatment. Clin Appl Thromb Haemost 2000;
6: 82-86. https://doi.org/10.1177/107602960000600206 PMid:10775027
- Solymoss, S. Postpartum acquired factor VIII inhibitors: results of a survey. Am J Hematol 1998; 59: 1-4. https://doi.org/10.1002/(SICI)1096-8652(199809)59:1<1::AID-AJH1>3.0.CO;2-T
- Franchini
M, Zaffanello M, Lippi G. Acquired hemophilia in pediatrics: a
systematic review. Pediatr Blood Cancer 2010;55 (October (4):606-11. https://doi.org/10.1002/pbc.22657 PMid:20589621
- Baudo
F, de Cataldo F. Acquired factor VIII inhibitors in pregnancy: data
from the Italian Haemophilia Registry relevant to clinical practice. Br
J Obstetr Gynecol 2003; 110: 311-314. https://doi.org/10.1046/j.1471-0528.2003.01535.x
- Tengborn
L, et al.; EACH2 registry contributors. Pregnancy-associated acquired
haemophilia A: results from the European Acquired Haemophilia (EACH2)
registry. Br J Obstet Gynecol 2012; 119: 1529-1537. https://doi.org/10.1111/j.1471-0528.2012.03469.x PMid:22901076
- Hauser,
I., Schneider, B. & Lechner, K. Post-partum factor VIII inhibitors.
A review of the literature with special reference to the value of
steroid and immunosuppressive treatment. Thrombosis and Haemostasis,
1995; 73: 1-5. https://doi.org/10.1055/s-0038-1651666 PMid:7740477
- Borg
JY, Guillet B, Le Cam-Duchez V, Goudemand J, Lévesque H; SACHA Study
Group.Haemophilia. 2013. Outcome of acquired haemophilia in France: the
prospective SACHA (Surveillance des Auto antiCorps au cours de
l'Hémophilie Acquise) registry. Haemophilia. 2013 Jul;19(4):564-570. https://doi.org/10.1111/hae.12138 PMid:23574453
- Tiede
A, Klamroth R, Scharf RE, Trappe RU, Holstein K, Huth-Kühne A,
Gottstein S, Geisen U, Schenk J, Scholz U, Schilling K, Neumeister P,
Miesbach W, Manner D, Greil R, von Auer C, Krause M, Leimkühler K,
Kalus U, Blumtritt JM, Werwitzke S, Budde E, Koch A, Knöbl P.
Prognostic factors for remission of and survival in acquired hemophilia
A (AHA): results from the GTH-AH 01/2010 study. Blood 2015 Feb
12;125(7):1091-1097. https://doi.org/10.1182/blood-2014-07-587089 PMid:25525118 PMCid:PMC4326770
- Huang
SY, Tsay W, Lin SY, Hsu SC, Hung MH, Shen MC. A study of 65 patients
with acquired hemophilia A in Taiwan. J Formos Med Assoc.
2015;114:321-327. https://doi.org/10.1016/j.jfma.2013.01.006 PMid:25839765
- Jayakar
JP, O'Neill N, Yan M, Nisenbaum R, Garvey MB, Teitel J, Sholzberg M.
Retrospective review of Acquired Haemophilia A from the largest
Canadian Haemophilia treatment centre. Haemophilia. 2018
Sep;24(5):e383-e387 https://doi.org/10.1111/hae.13598 PMid:30112783
- Wermes
C, Niekrens C, Sykora KW. Successful long-time treatment with
mycophenolate-mofetil in a child with acquired factor VIII inhibitor.
Hamostaseologie. 2012;32 Suppl 1: S75-8. https://doi.org/10.1055/s-0037-1619780
- Somaratne
PD, Jansan J, Senanayake HM, Ratnamalala V, Jayathilake MM,
Thirumavalavan K. A child with acquired haemophilia. Ceylon Med J. 2014
Jun;59 (2):66-67. https://doi.org/10.4038/cmj.v59i2.7068 PMid:24977427
- Fletcher
M, Crombet O, Morales-Arias J. Successful treatment of acquired
hemophilia a with rituximab and steroids in a 5-year-old girl. J
Pediatr Hematol Oncol. 2014 Mar;36(2):e103-104. https://doi.org/10.1097/MPH.0b013e318286d536 PMid:23588328
- Todo
K, Ohmae T, Osamura T, Kiyosawa N, Sugimoto M, Shima M, Imamura T,
Imashuku S. Exsanguinating bleeding following tooth extraction in a
12-year-old girl: a rare case of acquired haemophilia A. Blood Coagul
Fibrinolysis. 2015 Dec;26(8):964-966. https://doi.org/10.1097/MBC.0000000000000355 PMid:26397882
- Jones
L, Dandoy C, Jodele S, Myers KC, Luchtman-Jones L, Quinn CT, Mullins E,
El-Bietar J.Successful management of concurrent acquired hemophilia A
and a lupus anticoagulant in a pediatric hematopoietic stem cell
transplant patient. Bone Marrow Transplant. 2018 Apr;53(4):487-489. https://doi.org/10.1038/s41409-017-0041-0 PMid:29330401
- Takeyama
M, Nogami K, Kajimoto T, Ogiwara K, Matsumoto T, Shima M. First report
of real-time monitoring of coagulation function potential and IgG
subtype of anti-FVIII autoantibodies in a child with acquired
hemophilia A associated with streptococcal infection and amoxicillin.
Int J Hematol. 2018 Jan;107(1):112-116 https://doi.org/10.1007/s12185-017-2273-6 PMid:28597369
- Gamage
M, Weerasinghe S, Nasoor M, Karunarathne AMPW, Abeyrathne SP.
Progressive Intramuscular Haematoma in a 12-Year-Old Boy: A Case of
Acquired Haemophilia A. Case Rep Hematol. 2018 Oct 24; 2018:6208597 https://doi.org/10.1155/2018/6208597 PMid:30473893 PMCid:PMC6220402
- Godaert
L, Bartholet S, Colas S, Kanagaratnam L, Fanon JL, Dramé M. Acquired
Hemophilia A in Aged People: A Systematic Review of Case Reports and
Case Series. Semin Hematol. 2018 Oct;55(4):197-201 https://doi.org/10.1053/j.seminhematol.2018.02.004 PMid:30502847
- Yamaguchi
T, Kudo N, Endo S, Usui T, Imashuku S. Management of Acquired
Hemophilia A in Elderly Patients. Case Rep Hematol. 2018 Nov 13;
2018:6757345 https://doi.org/10.1155/2018/6757345 PMid:30538871 PMCid:PMC6260550
- Mudad R, Kane WH. DDAVP in acquired haemophilia A: case report and review of the literature. Am J Hematol 1993; 43: 295-299. https://doi.org/10.1002/ajh.2830430413 PMid:8372811
- Franchini M, Lippi G. The use of desmopressin in acquired haemophilia A: a systematic review. Blood Transfus 2011; 9: 377-382
- Baudo
F, et al.; EACH2 registry contributors. Management of bleeding in
acquired haemophilia A: results from the European Acquired Haemophilia
(EACH2) Registry. Blood 2012; 120: 39-46 https://doi.org/10.1182/blood-2012-02-408930 PMid:22618709
- Morrison
AE, et al. Use of porcine factor VIII in the treatment of patients with
acquired haemophilia. Blood 1993; 81: 1513-1520. https://doi.org/10.1182/blood.V81.6.1513.1513 PMid:8453098
- Giangrande PL. Porcine factor VIII. Haemophilia 2012 May;18(3):305-309 https://doi.org/10.1111/j.1365-2516.2012.02803.x PMid:22531020
- Doering
CB, Healey JF, Parker ET, Barrow RT, Lollar P. High level expression of
recombinant porcine coagulation factor VIII. J Biol Chem 2002; 277:
38345-9. https://doi.org/10.1074/jbc.M206959200 PMid:12138172
- Kempton
CL, Abshire TC, Deveras RA et al. Pharmacokinetics and safety of OBI-1,
a recombinant B domain-deleted porcine factor VIII, in subjects with
haemophilia A. Haemophilia 2012; 18: 798-804. https://doi.org/10.1111/j.1365-2516.2012.02789.x PMid:22512291
- Lillicrap
D., Schiviz A. , Apostol C., Wojciechowski F., Horling F. , Lai C. K. ,
Piskernik C., Hoellriegl W., and Lollar P. Porcine recombinant factor
VIII (Obizur; OBI-1; BAX801):product characteristics and preclinical
profile. Haemophilia. 2016 Mar; 22(2):308-317. https://doi.org/10.1111/hae.12784 PMid:26278557
- Kruse-Jarres
R, St-Louis J, Greist A, et al. Efficacy and safety of OBI-1, an
antihaemophilic factor VIII (recombinant), porcine sequence, in
subjects with acquired haemophilia A. Haemophilia 2015; 21:162-170. https://doi.org/10.1111/hae.12627 PMid:25623166
- Martin
K, Kasthuri R, Mooberry MJ, Chen SL, Key NS, Ma AD. Lower doses of
recombinant porcine factor VIII maintain excellent haemostatic
efficacy. Haemophilia. 2016 Nov; 22(6):e549-e551. https://doi.org/10.1111/hae.13038 PMid:27704655 PMCid:PMC5119759
- Tarantino
MD, Cuker A, Hardesty B, Roberts JC, Sholzberg M. Recombinant porcine
sequence factor VIII (rpFVIII) for acquired haemophilia A: practical
clinical experience of its use in seven patients. Haemophilia. 2017
Jan; 23(1):25-32. https://doi.org/10.1111/hae.13040 PMid:27511890
- Hay
CRM, Negrier C, Ludlam CA. The treatment of bleeding in acquired
haemophilia with recombinant factor VIIa: a multicenter study. Thromb
Haemost 1997; 78: 3-7. https://doi.org/10.1055/s-0038-1665434
- Baudo
F, de Cataldo F, Gaidano G; Italian registry of acquired hemophilia.
Treatment of acquired factor VIII inhibitor with recombinant activated
factor VIIa: data from the Italian registry of acquired hemophilia.
Haematologica. 2004 Jun;89(6):759-761.
- Sumner
MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of acquired
haemophilia with recombinant activated FVII: a critical appraisal.
Haemophilia 2007; 13: 451-461. https://doi.org/10.1111/j.1365-2516.2007.01474.x PMid:17880429
- Amano
K, Seita I, Higasa S, Sawada A, Kuwahara M, Shima M. Treatment of acute
bleeding in acquired haemophilia A with recombinant activated factor
VII: analysis of 10-year Japanese post-marketing surveillance data.
Haemophilia. 2017 Jan;23(1):50-58. https://doi.org/10.1111/hae.13033 PMid:27457022
- Franchini
M, Lippi G. Recombinant activated factor VII: Mechanisms of action and
current indications. Semin Thromb Haemost 2010; 36: 485-492. https://doi.org/10.1055/s-0030-1255442 PMid:20632246
- Sallah S. Treatment of acquired haemophilia with factor eight inhibitor by-passing activity. Haemophilia 2004; 10: 169-173. https://doi.org/10.1046/j.1365-2516.2003.00856.x PMid:14962206
- Borg
JY, Négrier C, Durieu I, Dolimier E, Masquelier AM, Lévesque H; FEIBHAC
Study Group.FEIBA in the treatment of acquired haemophilia A: results
from the prospective multicentre French 'FEIBA dans l'hémophilie A
acquise' (FEIBHAC) registry.Haemophilia. 2015 May;21(3):330-337 https://doi.org/10.1111/hae.12574 PMid:25359571
- Zanon
E, Pasca S, Santoro C, Gamba G, Siragusa SM, Rocino A, Cantori I,
Federici AB, Mameli L, Giuffrida G, Falanga A, Lodigiani C, Santoro RC,
Milan M, Ambaglio C, Napolitano M, Mazzucconi MG. Activated prothrombin
complex concentrate (FEIBA®) in acquired haemophilia A: a large
multicentre Italian study - the FAIR Registry. Br J Haematol. 2019
Mar;184(5):853-855. https://doi.org/10.1111/bjh.15175 PMid:29528100
- Ingerslev
J, Sorensen B. Parallel use of by-passing agents in haemophilia with
inhibitors: a critical review. Br J Haematol. 2011;155: 256-262. https://doi.org/10.1111/j.1365-2141.2011.08854.x PMid:21895627
- Pasca
S, Ambaglio C, Rocino A, Santoro C, Cantori I, Zanon E; FAIR Study
Group. Combined use of antifibrinolytics and activated prothrombin
complex concentrate (aPCC) is not related to thromboembolic events in
patients with acquired haemophilia A: data from FAIR Registry. J Thromb
Thrombolysis. 2019 Jan;47(1):129-133. https://doi.org/10.1007/s11239-018-1750-y PMid:30267246
- Kim
CH, Simmons SC, Bui CM, Jiang N, Pham HP. aPCC vs. rFVIIa for the
treatment of bleeding in patients with acquired haemophilia - a
cost-effectiveness model. Vox Sang. 2019 Jan;114 (1):63-72. https://doi.org/10.1111/vox.12726 PMid:30499154
- Franchini
M, et al. Extracorporeal immunoadsorption for the treatment of
coagulation inhibitors. Semin Thromb Haemost 2009; 35: 76-80. https://doi.org/10.1055/s-0029-1214150 PMid:19308895
- Oldenburg
J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, Santagostino
E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy
GG, Windyga J, Shima M. Emicizumab prophylaxis in hemophilia A with
inhibitors. N Engl J Med 2017; 377: 809-818. https://doi.org/10.1056/NEJMoa1703068 PMid:28691557
- Young
G, Liesner R, Chang T, Sidonio R, Oldenburg J, Jiménez-Yuste V,
Mahlangu J, Kruse-Jarres R, Wang M, Uguen M, Doral MY, Wright LY,
Schmitt C, Levy GG, Shima M, Mancuso ME. A multicenter, open-label
phase 3 study of emicizumab prophylaxis in children with hemophilia A
with inhibitors. Blood. 2019 Dec 12;134(24):2127-2138. https://doi.org/10.1182/blood.2019001869 PMid:31697801 PMCid:PMC6908828
- Mahlangu
J, Oldenburg J, Paz-Priel I, Negrier C, Niggli M, Mancuso ME, Schmitt
C, Jiménez-Yuste V, Kempton C, Dhalluin C, Callaghan MU, Bujan W, Shima
M, Adamkewicz JI, Asikanius E, Levy GG, Kruse-Jarres R. Emicizumab
Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N
Engl J Med. 2018 Aug 30;379(9):811-822. https://doi.org/10.1056/NEJMoa1803550 PMid:30157389
- Pipe
SW, Shima M, Lehle M, Shapiro A, Chebon S, Fukutake K, Key NS, Portron
A, Schmitt C, Podolak-Dawidziak M, Selak Bienz N, Hermans C,
Campinha-Bacote A, Kiialainen A, Peerlinck K, Levy GG, Jiménez-Yuste V.
Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given
every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre,
open-label, non-randomised phase 3 study. Lancet Haematol. 2019
Jun;6(6):e295-e305. https://doi.org/10.1016/S2352-3026(19)30054-7
- Knoebl
P, Sperr W, Schellongowski P, Staudinger T, Jilma‐Stohlawetz P,
Quehenberger P, et al. Emicizumab for the treatment of acquired
hemophilia A: lessons learned from 4 very different cases [abstract]
Blood. 2018;132(Suppl 1):2476. https://doi.org/10.1182/blood-2018-99-116973
- Dane
KE, Lindsley JP, Streiff MB, Moliterno AR, Khalid MK, Shanbhag S.
Successful use of emicizumab in a patient with refractory acquired
hemophilia A and acute coronary syndrome requiring percutaneous
coronary intervention. Res Pract Thromb Haemost. 2019 Apr
9;3(3):420-423. https://doi.org/10.1002/rth2.12201 PMid:31294330 PMCid:PMC6611359
- Möhnle
P, Pekrul I, Spannagl M, Sturm A, Singh D, Dechant C. Emicizumab in the
Treatment of Acquired Haemophilia: A Case Report. Transfus Med
Hemother. 2019 Apr;46(2):121-123. https://doi.org/10.1159/000497287 PMid:31191199 PMCid:PMC6514512
- Al-Banaa
K, Alhillan A, Hawa F, Mahmood R, Zaki A, El Abdallah M, Zimmerman J,
Musa F. Emicizumab Use in Treatment of Acquired Hemophilia A: A Case
Report. Am J Case Rep. 2019 Jul 18;20:1046-1048. https://doi.org/10.12659/AJCR.916783 PMid:31318850 PMCid:PMC6659457
- Takeyama M, Nogami K, Matsumoto T, Noguchi-Sasaki M, Kitazawa T, Shima M. An anti-factor IXa/factor X bispecific antibody,
emicizumab, improves ex vivo coagulant potentials in plasma from
patients with acquired hemophilia A. J Thromb Haemost. 2020 Jan 26.
doi: 10.1111/jth.14746. [Epub ahead of print] https://doi.org/10.1111/jth.14746 PMid:31984625
- Green
D, Rademaker AW, Briët E. A prospective, randomised trial of prednisone
and cyclophosphamide in the treatment of patients with factor VIII
autoantibodies. Thromb Haemost. 1993 Nov 15;70(5):753-757. https://doi.org/10.1055/s-0038-1649664 PMid:8128430
- Bitting
RL, Bent S, Li Y, Kohlwes J. The prognosis and treatment of acquired
hemophilia: a systematic review and meta-analysis. Blood Coagul
Fibrinolysis. 2009 Oct;20(7):517-523 https://doi.org/10.1097/MBC.0b013e32832ca388 PMid:19644360
- Collins
P, Baudo F, Knoebl P, Lévesque H, Nemes L, Pellegrini F, Marco P,
Tengborn L, Huth-Kühne A; EACH2 registry collaborators.
Immunosuppression for acquired hemophilia A: results from the European
Acquired Haemophilia Registry (EACH2). Blood. 2012 Jul 5;120(1):47-55. https://doi.org/10.1182/blood-2012-02-409185 PMid:22517903 PMCid:PMC3390961
- Nemes L, Pitlik E. New protocol for immune tolerance induction in acquired haemophilia. Haematologica 2000; 85: 64-68.
- Zeitler
H, Ulrich-Merzenich G, Hess L, Konsek E, Unkrig C, Walger P, Vetter H,
Brackmann HH. Treatment of acquired haemophilia by the Bonn-Malmo
Protocol: documentation of an in vivo immunomodulating concept. Blood
2005; 105:2287-2293. https://doi.org/10.1182/blood-2004-05-1811 PMid:15542586
- Franchini
M, Mannucci PM. Inhibitor eradication with rituximab in haemophilia:
where do we stand? Br J Haematol. 2014 Jun;165(5):600-608. https://doi.org/10.1111/bjh.12829 PMid:24628543
- D'Arena
G, Grandone E, Di Minno MN, Musto P, Di Minno G. The anti-CD20
monoclonal antibody rituximab to treat acquired haemophilia A. Blood
Transfus. 2016 May;14(2):255-261
- Maillard
H, Launay D, Hachulla E, Goudemand J, Lambert M, Morell-Dubois S,
Queyrel V, Hatron PY. Rituximab in postpartum-related acquired
hemophilia. Am J Med. 2006 Jan;119(1):86-88. https://doi.org/10.1016/j.amjmed.2005.06.068 PMid:16431202
- Santoro
C, Rago A, Biondo F, De Propris MS, De Vellis A, Guarini A, Pignoloni
P, Mazzucconi MG. Efficacy of rituximab treatment in post-partum
acquired haemophilia A. Haemophilia. 2008 Jan;14(1):147-149.
- Dedeken
L, St-Louis J, Demers C, Meilleur C, Rivard GE. Postpartum acquired
haemophilia: a single centre experience with rituximab. Haemophilia.
2009;15:1166-1168 https://doi.org/10.1111/j.1365-2516.2009.02008.x PMid:19500171
- Kyrle PA. High factor VIII and the risk of venous thromboembolism. Hamostaseologie 2003;23:41-44 https://doi.org/10.1055/s-0037-1619564 PMid:12567199
- Mizrahi
T, Doyon K, Dubé E, Bonnefoy A, Warner M, Cloutier S, Demers C,
Castilloux JF, Rivard GE, St-Louis J. Relapse pattern and long-term
outcomes in subjects with acquired haemophilia A. Haemophilia. 2019
Mar;25(2):252-257. https://doi.org/10.1111/hae.13685 PMid:30694571
- Huth-Kühne
A, Baudo F, Collins P, Ingerslev J, Kessler CM, Lévesque H, Mingot
Castellano ME, Shima M, and St-Louis J. International recommendations
on the diagnosis and treatment of patients with acquired hemophilia A.
Haematologica 2009; 94:566-575 https://doi.org/10.3324/haematol.2008.001743 PMid:19336751 PMCid:PMC2663620
- Franchini
M, Castaman G, Coppola A, Santoro C, Zanon E, Di Minno G, Morfini M,
Santagostino E, Rocino A, on behalf of the AICE Working Group. Acquired
inhibitors of clotting factors: AICE recommendations for diagnosis and
management. Blood Transfus 2015; 13; 498-513
- Tiede
A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, Di Minno G,
d'Oiron R, Salaj P, Jiménez-Yuste V, Huth-Kühne A,and Paul Giangrande.
International recommendations on the diagnosis and treatment of
acquired hemophilia A. Haematologica. 2020; 105:xxx
doi:10.3324/haematol.2019.230771 https://doi.org/10.3324/haematol.2019.230771 PMid:32381574
- Guyatt
GH, Oxman AD, Kunz R, Vist GE, Falck-Ytter Y, Schünemann HJ; GRADE
Working Group. What is "quality of evidence" and why is it important to
clinicians? BMJ 2008; 336: 995-998. https://doi.org/10.1136/bmj.39490.551019.BE PMid:18456631 PMCid:PMC236480
[TOP]