Johanna Flach1*, Evgenii Shumilov2*, Naomi Porret3,4, Inna Shakhanova5, Myriam Legros4, Marie-Noëlle Kronig6, Raphael Joncourt3,4, Ulrike Bacher3,4** and Thomas Pabst6**.
1 Department of Hematology and Oncology, Medical Faculty Mannheim of the Heidelberg University, Mannheim, Germany.
2 Department of Hematology and Medical Oncology, University Medicine Göttingen (UMG), Göttingen, Germany.
3
University Department of Hematology and Central Hematology Laboratory,
Inselspital, Bern University Hospital, Bern, Switzerland.
4
Center of Laboratory Medicine (ZLM)/University Institute of Clinical
Chemistry, Inselspital, Bern University Hospital, Bern, Switzerland.
5 Department of Nephrology and Rheumatology, University Medicine Göttingen (UMG), Göttingen, Germany.
6 Department of Medical Oncology, Inselspital, Bern University Hospital, Bern, Switzerland.
Correspondence to: Ulrike Bacher, MD. Department of Hematology and
Center of Laboratory Medicine (ZLM); Inselspital, Bern University
Hospital, University of Bern; Bern, Switzerland; Tel. +41-31-632-1390;
Fax. +41-31-632-3406. E-mail:
veraulrike.bacher@insel.ch Thomas
Pabst, MD. Department of Medical Oncology; Inselspital, Bern University
Hospital, University of Bern; Bern, Switzerland; Tel.: +41-31-632 8430;
Fax: +41-31-632-3410. E-mail:
thomas.pabst@insel.ch
Published: September 1, 2020
Received: June 28, 2020
Accepted: August 16, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020068 DOI
10.4084/MJHID.2020.068
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
To the editor,
Next-generation
sequencing (NGS) has opened the opportunity for assessing the unique
clonal composition of patients with acute myeloid leukemia (AML), not
only at the beginning but also within the course of the disease, and
particularly at relapse. The molecular genetic characterization of the
clonal composition has therapeutic relevance, as a number of
molecularly directed treatment options have recently become available.
Since relapse of AML remains a major clinical challenge, comprehensive
diagnostics during follow-up and at relapse has become an increasingly
important pillar of clinical decision making. In the current
manuscript, we have applied myeloid NGS panel sequencing to compare the
genetic profiles of six illustrative AML patients at initial diagnosis
and at relapse. We found that NGS has the potential to identify clonal
molecular stability, evolution, and devolution in addition to
co-occurring changes on the cytogenetic level, all of which can occur
alone or in combination. We discuss these patients in detail, covering
clinical, molecular, and cytogenetic, as well as therapeutic aspects.
The
increasing use of NGS has been enabled by a number of commercially
available panels that cover the most frequently mutated genes. In
contrast to traditional diagnostic tools (i.e., cytomorphology,
cytogenetics, qPCR) that provide classification and prognostic
information only within certain categories, molecular profiling by NGS
enables us to depict a unique genetic make-up for each AML patient.
Genetic information provides crucial parameters within the current AML
classification systems[1,2] and has not only an impact on prognosis but
also influences treatment options.
However, despite improving
remission rates, around 40-60% of AML patients will ultimately
relapse,[3] which remains the major determinant of outcome. At relapse,
AML patients can either present with the same genetic mutation pattern
as observed at initial diagnosis (clonal stability), or present with
higher complexity, e.g., through the acquisition of additional
mutations (clonal evolution), or lose some of the initial mutations at
relapse (clonal devolution), or, alternatively, show both gains and
losses of mutations.[4-6]
Concerning the pathophysiologic
mechanisms involved during clonal evolution at relapse, our current
paradigms suggest that pre-existing clones (or subclones) may gain a
survival advantage under the selective chemotherapeutic pressure.[6] We
have recently provided a comprehensive overview of the current
knowledge of NGS during relapse of AML.[7] Here, we present six
illustrative patient studies observed during clinical practice to
demonstrate characteristic genetic scenarios accompanying hematologic
relapse of AML following intensive chemotherapy. All patients were
treated and analyzed by cytogenetics and myeloid NGS panels at
diagnosis and at relapse at our department according to the methodology
described in Table 1. The
sensitivity of NGS analyses was limited to a 5% variant allele
frequency (VAF) at diagnosis, and 1% at follow-up as the exact mutation
localizations were known.
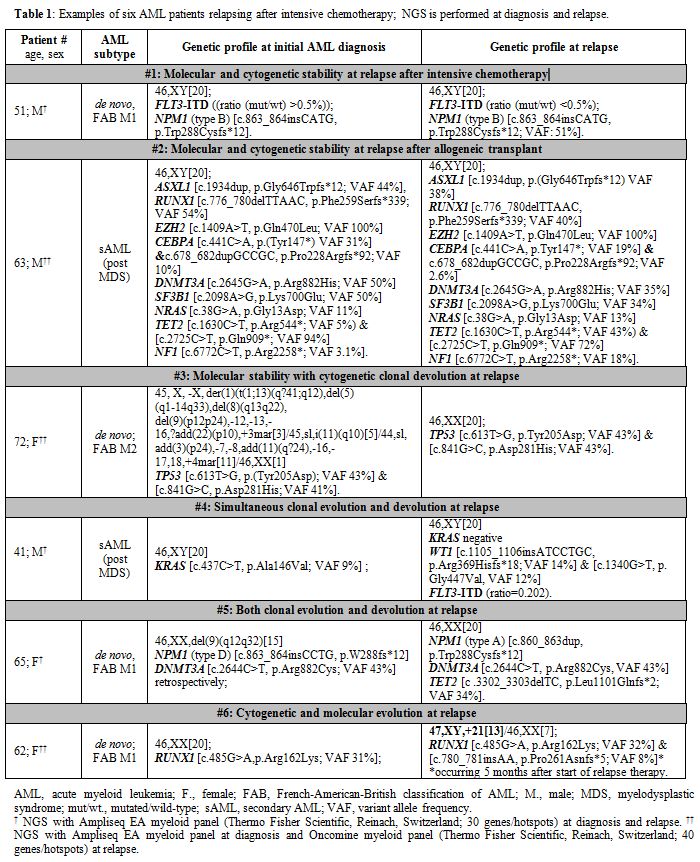 |
Table
1. Examples of six AML patients relapsing after intensive chemotherapy; NGS is performed at diagnosis and relapse. |
Patient #1: Molecular and cytogenetic stability at relapse after intensive chemotherapy
A 51-year-old male patient presented with de novo AML, FAB M1, 46, XY, and bone marrow (BM) blasts >90%. Fragment analysis revealed an FLT3-ITD with a mutant/wild-type ratio of >0.5, and PCR detected an NPM1
(type B) mutation. No additional mutations were detected by NGS.
Standard induction chemotherapy (cytarabine/ idarubicin followed by
cytarabine/daunorubicin) induced complete morphologic remission (CR)
with minimal/measurable residual disease (MRD) positivity according to
HOVON-132 for the previously known NPM1
mutation. After high-dose chemotherapy (HDCT) with
busulfan/cyclophosphamide and autologous stem cell transplantation
(ASCT), the patient achieved molecular MRD-negative CR1 as assessed by
PCR/fragment analysis. 4.5 months after ASCT, both the NPM1 and FLT3-ITD
mutations re-emerged, followed by an overt hematologic relapse one
month later. NGS and cytogenetics revealed no additional changes.
Salvage therapy (cladribine, cytarabine, and idarubicin; CLA-Ida) plus
sorafenib resulted in morphologic CR2, albeit with the persistence of
the NPM1 and FLT3-ITD
mutations. Matched-related myeloablative allogeneic HSCT (11/2017) was
performed, but molecular MRD persisted at 3 months post-transplant, and
sorafenib was once more initiated and combined with azacitidine (AZA).
At the time of this report, the patient remained in molecular CR2,
meanwhile under continued sorafenib monotherapy.
Patient #2: Molecular and cytogenetic stability at relapse after allogeneic transplant
A
63 years old male patient developed s-AML following 4 years of MDS with
a “wait and watch” approach. At the time of s-AML, the karyotype was
normal, and NGS revealed nine mutations in ASXL1, RUNX1, EZH2, CEBPA, DNMT3A, SF3B1, NRAS, TET2, and NF1.
The patient underwent cytarabine/idarubicin induction followed by
myeloablative allogeneic HSCT in CR1 with the molecular persistence of
all mutations. Subsequently, the patient developed hematologic relapse
(d+78 after HSCT), demonstrating a normal karyotype and the identical
mutations seen before. The patient was resistant to AZA, donor
lymphocyte infusion, and gemtuzumab ozogamicin and died 4 months after
HSCT due to progressive disease.
Patient #3: Molecular stability with cytogenetic clonal devolution at relapse
A 72 years old female patient was diagnosed with high-risk de novo AML M2 with a complex multi-clonal karyotype. NGS revealed two different mutations in TP53.
The patient was refractory to standard induction chemotherapy, but
second-line decitabine (10 cycles) resulted in CR1, and NGS documented
clearance of the TP53
mutations. Due to poor veins, decitabine therapy was switched to AZA.
Relapse occurred 11.5 months after the achievement of CR1; the
karyotype was normal, whereas both TP53
mutations remained detectable. Decitabine treatment combined with
sorafenib failed to induce any response. The patient died one month
after relapse detection due to progressive disease.
Patient #4: Simultaneous clonal evolution and devolution at relapse
Forty-one
years old male patient presented with s-AML transformed from untreated
high-risk MDS diagnosed three months earlier. Genetic analysis revealed
a normal karyotype and an isolated KRAS
mutation by NGS. Induction with two cycles of cytarabine and idarubicin
resulted in MRD-negative CR1. ASCT was performed after
melphalan/cyclophosphamide HDCT, but hematologic relapse occurred 6.4
months following ASCT. In contrast to the results obtained in the clone
at initial diagnosis, the BM at relapse was negative for the KRAS mutation but presented with two WT1
mutations and FLT3-ITD (ratio=0.202). The patient received CLA-Ida
salvage therapy and allogeneic HSCT from his HLA-identical sister. He
has been in hematologic and molecular CR2 at last follow-up 19 months
after relapse.
Patient #5: Both clonal evolution and devolution at relapse
A 65 years old female patient presented with de novo AML, FAB M1, with a 9q deletion. PCR revealed NPM1
mutation type D. The patient received induction consisting of
cytarabine/idarubicin/laromustin followed by busulfan/cyclophosphamide
HDCT/ASCT. Following a period of long-lasting remission with MRD
negativity for the NPM1
type D mutation over 8 years, the patient showed a 4-log increase of
the NPM1 mutation load detected by qPCR. Still being at hematologic
CR1, NPM1
re-appearance was detected by qPCR as well as NGS but surprisingly
identified type A instead of type D. Yet, the latter was also confirmed
retrospectively by NGS at initial diagnosis. In addition, a DNMT3A
mutation was discovered in the relapse sample. Subsequent qPCR assay
with a primer designed for type A revealed a ratio of 6.285. Six weeks
later, BM cytomorphology and immunophenotyping revealed up to 35%
myeloid blasts corresponding to overt AML M2. Cytogenetics revealed a
normal karyotype, with a lack of the 9q deletion documented at first
diagnosis. We have previously described this switch of NPM1 types (from type D to type A).[9] The DNMT3A
mutation identified at relapse was retrospectively detected in stored
material from initial diagnosis. The patient underwent salvage
chemotherapy, followed by busulfan/melphalan HDCT/ASCT. Subsequently,
maintenance therapy with AZA was started resulting in NPM1 MRD-negative CR2 ongoing 22 months after diagnosis of relapse. Finally, a TET2 mutation was also found retrospectively with a variant allele frequency (VAF) of 43% in addition to DNMT3A (47% VAF) in the autologous stem cell harvest preceding the 2nd ASCT. TET2 and DNMT3A mutations persisted at last follow-up (34%/43% VAF) despite NPM1 type A mutation clearance suggesting an interpretation of clonal hematopoiesis of indeterminate potential (CHIP).
Patient #6: Cytogenetic and molecular evolution at relapse
A 62-year-old female patient came to observation with de novo AML, FAB M1, and a peripheral blast count of 55%. The NGS panel revealed a RUNX1
mutation with a VAF of 31%, and cytogenetics showed a normal karyotype.
The patient received two cycles of induction (daunorubicin,
cytarabine), resulting in CR1 and MRD-negativity for the RUNX1
mutation by NGS. This was followed by HDCT consisting of
busulfan/melphalan and ASCT since the patient declined allogeneic
transplantation. Hematological regeneration was heavily delayed. Five
months after ASCT, the patient developed hematologic relapse. At this
stage, BM presented a clonal cytogenetic evolution with a novel gain of
chromosome 21 in addition to the re-emergence of the RUNX1
mutation that had already been present at the initial diagnosis.
Relapse therapy was initiated by decitabine after the patient declined
intensive re-induction treatment. Blasts, however, persisted, which was
5 months later accompanied by an additional, i.e., second, RUNX1
mutation suggesting further molecular clonal evolution. Palliative
therapy was administered by decitabine (9 cycles), then with low-dose
cytarabine with glasdegib (hedgehog pathway inhibitor), and finally,
sorafenib. The patient succumbed to refractory disease one month after
the sorafenib start and 15 months after the first relapse.
Conclusions
As
demonstrated by our case series, each patient may present with an
individual genetic composition at relapse of AML. Compared to the
initial diagnosis, this may comprise clonal stability, evolution, or
devolution alone or in combination both at the molecular and/or
cytogenetic level. Consequently, the genetic characterization during
relapse may identify novel lesions treatable by targeted therapies or
may open new pathways for bridging strategies towards allogeneic HSCT.
This can be illustrated, for example, by FLT3 mutations, which are emerging in around 10% of AML patients at relapse,[10] and may provide an option for specific FLT3 inhibitor treatment, such as midostaurin, gilteritinib or others.[11,12]
Similarly, both IDH1 (ivosidenib) and IDH2 (enasidenib) inhibitors were
recently approved in AML with the respective mutations.[13,14] Knowledge of relapse genetics may imply consequences also for prognosis. Adverse prognostic markers, such as TP53
mutations emerging at relapse, may allow timely initiation of donor
search for subsequent allogeneic HSCT. Accordingly, the development of
distinct diagnostics and therapeutic algorithms for clonal stability,
evolution, and devolution, as well as defining of “founder” mutations
in relapsed AML settings may further ease the management of such
patients. Anticipating anti-relapse treatment with targeted agents, the
determination of mutant allele frequencies is of high importance as
these provide a sensitive diagnostic tool to assess response on the
molecular level and predict progression over time. In conclusion, NGS
may be discussed for all patients at AML relapse. Due to recent
improvements in treatment options and an increasing understanding of
the molecular drivers of AML, therapy in the relapse situation becomes
more and more individualized, and, consequently, NGS will gain
increasing importance in this scenario.
References
- Arber DA, Orazi A, Hasserjian R, et al. The 2016
revision to the World Health Organization classification of myeloid
neoplasms and acute leukemia. Blood 2016;127:2391-2405. https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Dohner
H, Estey E, Grimwade D, et al. Diagnosis and management of AML in
adults: 2017 ELN recommendations from an international expert panel.
Blood 2017;129:424-447. https://doi.org/10.1182/blood-2016-08-733196 PMid:27895058 PMCid:PMC5291965
- Burnett
AK, Goldstone A, Hills RK, et al. Curability of patients with acute
myeloid leukemia who did not undergo transplantation in first
remission. J Clin Oncol 2013;31:1293-1301. https://doi.org/10.1200/JCO.2011.40.5977 PMid:23439754
- Greif
PA, Hartmann L, Vosberg S, et al. Evolution of Cytogenetically Normal
Acute Myeloid Leukemia During Therapy and Relapse: An Exome Sequencing
Study of 50 Patients. Clin Cancer Res 2018;24:1716-1726. https://doi.org/10.1158/1078-0432.CCR-17-2344 PMid:29330206
- Quek
L, Ferguson P, Metzner M, et al. Mutational analysis of disease relapse
in patients allografted for acute myeloid leukemia. Blood Adv
2016;1:193-204. https://doi.org/10.1182/bloodadvances.2016000760 PMid:29296935 PMCid:PMC5737177
- Wong
TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin
and evolution of therapy-related acute myeloid leukaemia. Nature
2015;518:552-555. https://doi.org/10.1038/nature13968 PMid:25487151 PMCid:PMC4403236
- Flach
J, Shumilov E., Wiedemann, G., et al. Clinical potential of introducing
next-generation sequencing in patients at relapse of acute myeloid
leukemia. Hematol Oncol 2020;1-7. https://doi.org/10.1002/hon.2739 PMid:32306411
- Shumilov
E, Flach J, Joncourt R, et al. Clinical value of molecular MRD
monitoring by next-generation sequencing in patients with IDH2 mutated
AML. Leuk Lymphoma 2019;60:2588-2590. https://doi.org/10.1080/10428194.2019.1585838 PMid:30998101
- Bacher
U, Porret N, Joncourt R, et al. Pitfalls in the molecular follow up of
NPM1 mutant acute myeloid leukemia. Haematologica 2018;103:e486-e488. https://doi.org/10.3324/haematol.2018.192104 PMid:29903758 PMCid:PMC6165800
- McCormick
SR, McCormick MJ, Grutkoski PS, et al. FLT3 mutations at diagnosis and
relapse in acute myeloid leukemia: cytogenetic and pathologic
correlations, including cuplike blast morphology. Arch Pathol Lab Med
2010;134:1143-1151.
- Perl AE, Martinelli
G, Cortes JE, et al. Gilteritinib or Chemotherapy for Relapsed or
Refractory FLT3-Mutated AML. N Engl J Med 2019;381:1728-1740. https://doi.org/10.1056/NEJMoa1902688 PMid:31665578
- Stone
RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for
Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med
2017;377:454-464. https://doi.org/10.1056/NEJMoa1614359 PMid:28644114 PMCid:PMC5754190
- Norsworthy
KJ, Luo L, Hsu V, et al. FDA Approval Summary: Ivosidenib for Relapsed
or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1
Mutation. Clin Cancer Res 2019;25:3205-3209. https://doi.org/10.1158/1078-0432.CCR-18-3749 PMid:30692099
- Stein
EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed
or refractory acute myeloid leukemia. Blood 2017;130:722-731. https://doi.org/10.1182/blood-2017-04-779405 PMid:28588020 PMCid:PMC5572791
[TOP]