Fernanda Cozendey Anselmo1,2, Abdou Gafar Soumanou2, Cleidiane de Aguiar Ferreira4, Flora Maia Viga Sobrinha4, Ana Caroline Santos Castro2, Rafael Oliveira Brito2, Adolfo José da Mota2, Marilda de Souza Gonçalves3 and José Pereira de Moura Neto1,2.
1 Universidade do Estado do Amazonas - Fundação Hospitalar de Hematologia e Hemoterapia do Amazonas, Manaus, Amazonas, Brasil.
2 Universidade Federal do Amazonas, Faculdade de Ciências Farmacêuticas, Manaus, Amazonas, Brasil.
3 Fundação Oswaldo Cruz - Centro de Pesquisas Gonçalo Moniz, Salvador, Bahia, Brasil.
4 Secretaria do Estado do Amazonas – SUSAM.
Published: January 1, 2021
Received: April 21, 2020
Accepted: December 7, 2020
Mediterr J Hematol Infect Dis 2021, 13(1): e2021001 DOI
10.4084/MJHID.2021.001
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Alpha Thalassemia (α-thal) is a heterogeneous group of hereditary
alterations caused by deletions that affect alpha regulatory genes, and
the 3.7Kb deletion is the most frequent worldwide. The prevalence
ranges from 20% and 35% in Brazil, depending mainly on race,
predominant in Afro-descendants.
Purpose: The aim was to determine α-thal -α3.7Kb and -α4.2Kb deletions, estimating their frequency in individuals from six regions of Amazonas State.
Methods:
Volunteers age between 18-59 years old of both genders participated in
the study. Blood was collected from March 2014 to September 2017 at the
health centers of each participant city. α-thal3.7Kb was performed by GAP-PCR, while α-thal4.2Kb
by Multiplex-PCR. The total samples collected from each city were:
Manaus (capital), 356 (19.7%); Iranduba 232 (12.8%); Manacapuru, 287
(15.9%); Presidente Figueiredo, 370 (20.5%); Itacoatiara, 301 (16.6%);
and Coari, 263 (14.5%).
Results:
The average age among males was 35.3±14.8, while for females, it was
36.7±14.9 years old. Microcytosis (MCV <80fL) was found in 158
individuals (8,46%) and α-thal diagnosed in 143 individuals (7.9%), and
all of these individuals carried the 3.7Kb deletion 5.95% in heterozygous and 1.95% in homozygous. α-thal4.2kb was not found in any volunteer. The association analyses to the α-thal3.7kb
genotypes were statistically significant for all hematological
parameters (p<.001), except serum iron and serum ferritin analyses.
Conclusion:
This study highlights α-thal 3.7kb deletion as an important public
health problem, especially in a population not yet characterized about
this disease. Thus, epidemiological studies using molecular tools
become relevant in regions where the disease is underestimated,
contributing to a better understanding of thalassemia incidence and
iron deficiency anemias incidence of the participating cities. We
reinforce that future molecular studies in North Region from Brazil can
be utilized to describe other genetic anemias as structural
hemoglobinopathies that have already proven to be highly prevalent in
Brazil.
|
Introduction
Alpha
thalassemia (α-thal) is characterized by the reduction or absence of
α-chain production. Widely distributed, alpha globin's chains deletion
is often found in tropical regions and is especially common in
Southeast Asia, the Indian Subcontinent, Africa, and the Middle East,
with frequencies rising from 70% to 90%.[1,2] The
degree of severity varies according to the number of involved genes and
may range from an individual asymptomatic to a life-incompatible
condition.[3]
The most common α-thal-1 forms found are --SEA, --FIL, --MED and --THAI. However, the most frequent form of deletion is the α-thal-2 (α+), that affects only one out of the four α-globin genes and whose alterations -α3.7Kb and -α4.2Kb are the most prevalent throughout the world.[4,5]
The
clinical severity of heterozygous carriers individuals is low; they
usually present milder symptoms; however, their red blood cell
deficiencies have to be differentiated from subtle anemia,
microcytic-hypochromic anemia, refractory or iron deficient. On the
other hand, the homozygous form accompanies signs ranging from moderate
to severe forms, such as hemolytic anemia.[6,7]
Widely
distributed, the frequency of α-thal is directly linked to the world's
constant migratory waves in recent centuries. For example, Africans
were taken to North and South America during the European colonization,
or among Vietnamese refugees, or in the latest crisis in Syria - which
has sent about a million people from the Middle East to Europe and the
Americas through Turkey. All these individuals come from areas with a
high incidence of thalassemia, and consequently, there is a genetic
flux between the country of origin and country of destination. The new
genetic background can lead to thalassemia at all levels of the disease
and favor the shuffling of mutations that are not commonly seen in
their local population.[8,9]
The aim of this study was to determine the frequency of thalassemia alpha -α3.7Kb and -α4.2Kb
deletions in individuals living in Manaus, capital of the State of
Amazonas, and from cities within the metropolitan region of Manaus.
Besides, to characterize the hematological parameters, serum ferritin,
and serum iron to each population and evaluate its association.
Materials and Methods
The
studied population was composed by volunteers (> 18 years old),
naturals from the State of Amazonas, of both genders, from outpatient
units in six cities: Manaus (N=356); Coari (N=263); Manacapuru (N=287);
Iranduba (N=232); Presidente Figueiredo (N=370) and Itacoatiara
(N=301). All samples were recruited of the outpatients randomly at of
cities. Subjects under 18 years old, pregnant, transfused in the last
three months, and patients with onco-hematological and/or hospitalized
conditions were not included in this study.
A total of 1809
peripheral blood samples were collected in three years, from 2014 to
2017. The hematological analyses were performed at the respective
outpatient units of the respective study cities. These analyses were
performed in the automated hematology analyzers of the new generation
impedance technique and always calibrated before every test: BC-5800
(Mindray, Shenzhen, China), Pentra XL (ABX 80 Horiba®,
France), and ADVIA 120 Hematology (Siemens Healthineers Brasil). For
serum ferritin and serum iron analyses were used Bioclin® KIT by
immunoturbidimetry and colorimetric assays, respectively, carried out
in a Bioclin 3000 (Quibasa-Belo Horizonte, Brazil).
The genomic
DNA was prepared using the BIOPUR Mini Spin Plus® extraction kit,
following the manufacturer's recommendations. The integrity and DNA
quantification were evaluated by NanodropTM 2000 (Thermofisher®).
The α-thalassemia 3.7Kb deletion was executed as by a previous study,[10] and 4.2Kb deletion by Multiplex- PCR technique adapted from the previous study using only primers of wild type alpha genes and 4.2Kb deletion.[11]
The PCR products were submitted to electrophoresis (Bio-Rad, EUA) in
1.5% agarose gel and visualized under ultraviolet light in ENDURO™ GDS
Gel Documentation System (Labnet International, New York, USA).
This
project was approved by the Ethics in Research Committee (CEP) from
Universidade Federal do Amazonas and Fundação Hospitalar de Hematologia
e hemoterapia, based on the Brazil Platform in three projects: N°
834.086, CAEE 30668114.0.0000.5020; N° 213.167, CAAE:
01193312.4.0000.0009; and N° 1.178.117, CAAE: 46020315.4.0000.5020.
The
distribution of variables analysis was performed using the
Kolmogorov-Smirnov test. The parameter values were presented as mean
and standard deviation. The One Way ANOVA parametric test was used to
analyze the distribution of the means of quantitative variables with
normal distribution within categories. As independent variables, the
groups were divided into α-Thalassemia genotypes, gender, and cities.
As dependent variables, the continuous data were age in years,
Hematological parameters, and iron serum and ferritin values.
Contingency table chi-square tests were performed comparing the
incidence of α-thalassemia between Cities. p<0.05 was
considered significant. Statistical analyzes were performed using SPSS
version 19.0 (Chicago, EUA) and GraphPad Prism version 5.0
(San Diego, EUA) software.
Results
The
studied population was composed predominantly of females (N=1049, 58%),
against 760 (42%) males. The average age among females was 36.7±14.9
years old and 35.3±14.8 years old for males (Table 1).
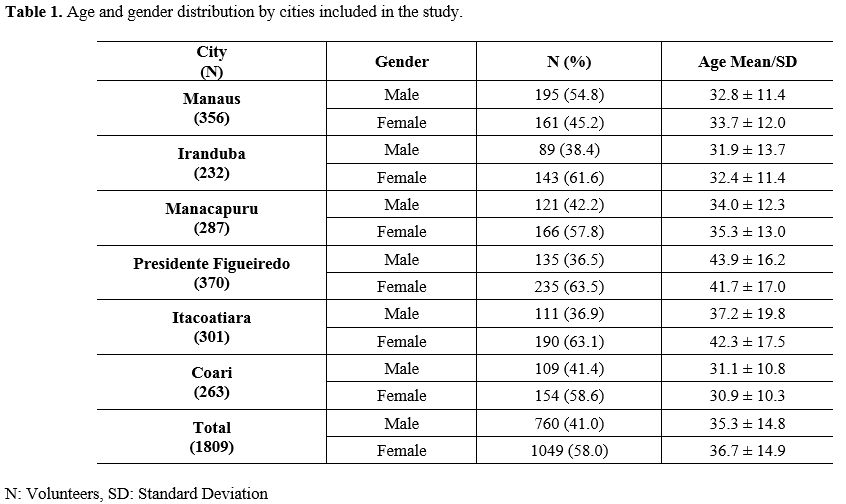 |
Table
1. Age and gender distribution by cities included in the study.
|
The alpha thalassemia screening found 143 individuals (7.9%) harboring the -α3.7Kb
deletion: 108 (6%) in heterozygous (-α/αα) and 35 (1.9%) in homozygous
(-α/-α). The prevalence in males was 7.9% (95% CI 6.0-9.9) and females
8.0% (CI 6.4-9.8) (Fisher test, p = 0.92). The frequency in Manaus was
7.9 (95% CI 5.1 - 10.7); Iranduba 7.3 (95% CI 3.9 - 10.8), Manacapuru
4.5% (95% CI 2.4 - 7.0), Presidente Figueiredo 10.3 (95% CI 7.3 -
13.2), Itacoatiara 9.6 (95% CI 6.3 - 13.3), Coari 7.2 (95% CI 4.2 -
10.3). The 4.2Kb deletion was not found in our studied population.
The
Leukocytes counts (WBC), erythrocytes (RBC), Hemoglobin (Hgb), Mean
Corpuscular Volume (MCV), Mean Corpuscular Hemoglobin (MCH), Mean
Corpuscular Hemoglobin Concentration (MCHC), Anisocytosis Index (RDW),
serum iron and serum ferritin were analyzed following stratification
for the α-thal genotype, i.e., one wild type group (αα/αα) (Supplementary Table 1A); one group composed only of heterozygous (-α/αα) individuals (Supplementary Table 1B); and one group composed only of homozygotes (-α/-α) individuals (Supplementary Table 1C).
When analyzing only wild types genotypes (αα/αα) between cities, the
association were statistically significant among all hematological
parameters, including for the serum iron and serum ferritin analyzes
between (p<0.001). Although statistically different (p <.001),
the hematological indexes and parameters for all cities are within the
normal reference values.[12]
Hematological
levels and iron test values were higher in men than women, according to
the literature. Hemoglobin, hematocrit, and erythrocytes values were
corrected for sex (Supplementary Table 2).
As expected, a higher frequency of a-thal was observed in those with microcytosis (40.68%), against 4.65% in normocytic (Figure 1).
We demonstrated that eight individuals have concomitantly α-thalassemia
and iron deficiency, representing 4% of the total number of α-thal
carriers.
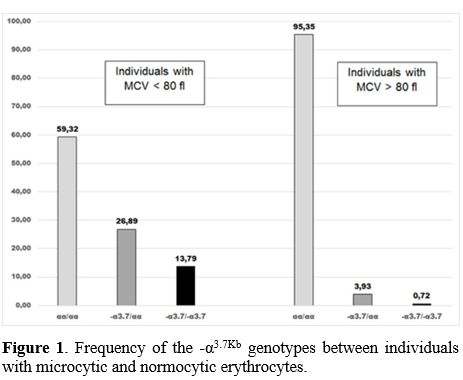 |
Figure
1. Frequency of the -α3.7Kb genotypes between individuals with microcytic and normocytic erythrocytes.
|
By the contingency
table chi-square tests, significant differences were found when
comparing the lowest and highest prevalence of a-thal with other
cities; Manacapuru vs. Presidente Figueiredo (p=.010 ), vs. Manaus
(p=.119), vs. Iranduba (p=.242), vs. Itacoatiara (p=.025), vs. Coari
(p=.332) and Presidente Figueiredo vs. Manaus (p=.318), vs. Iranduba
(p=.283), Itacoatiara (p=.886), Coari (p=.176).
Moreover, finally, we create a map of the -a3.7Kb frequencies found in the Amazon region. The results showed that -α3.7Kb
allele frequency was the highest in the Presidente Figueiredo City
(10.3%) located 98 km from Manaus and lowest followed in Itacoatiara
City (4.5%) located 128 km from Manaus (Figure 2).[13]
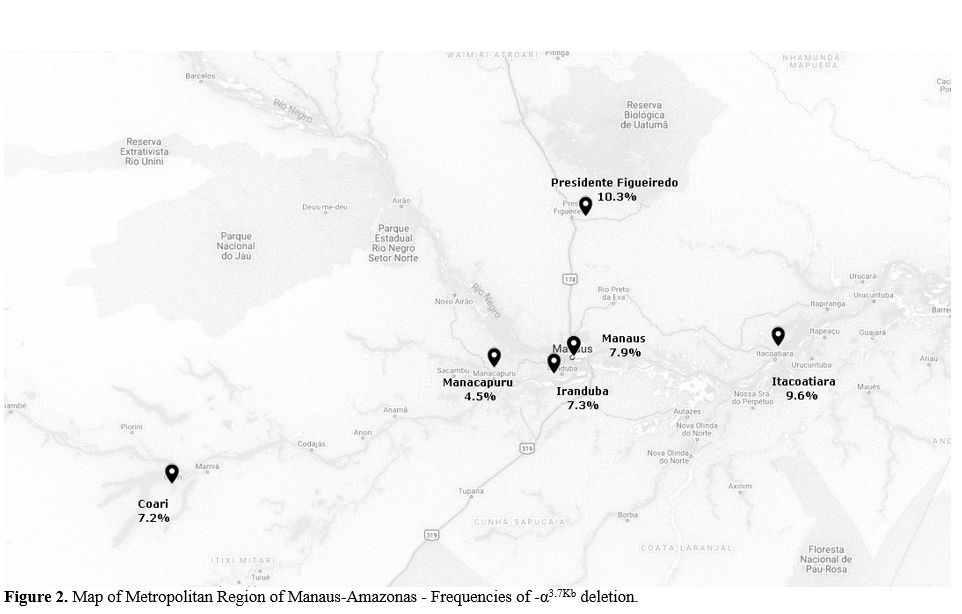 |
Figure 2. Map of Metropolitan Region of Manaus-Amazonas - Frequencies of -α3.7Kb deletion.
|
Discussion
The
α-thal occurrence in the city of Manaus-AM is still based only on
screening methods, such as hematological indices (MCV and MCH) and
supravital staining to detect Hb H inclusions (denatured β4 tetramers).
However, none of these approaches is entirely reliable or sensitive to
detect α-thalassemia trait (-α/αα). This problem is easily overcome by
the molecular tools applied to the alpha thalassemia's genotyping's
various genetic determinants.[14-16]
Although our group has already shown the 5.35% frequency -α3.7Kb is the deletion in blood donors from Manaus,[17]
this disease has never been studied in Manaus metropolitan region using
molecular methods. Thus its real prevalence is unknown and probably
underestimated.[18]
In this study, the -α3.7Kb deletion was uniquely found in 7.9% of our population, consistent with the high incidence in all States from Brazil.[19.20] In most studies, including Brazil, -α3.7Kb
is the deletion more frequently reported, ranging from 70% to 90% in
the regions of Melanesia and Nepal; reaching out 70.7% in Iran, 72.8%
in the Middle East, 35.2% in India, 16.3% in Thailand, 40% in African
countries and from 5 to 20% in Brazil.[21-25]
We
believed that our study shows some limitations since not all population
was included to make the prevalence estimation. However, we do not have
selection bias once individuals were free to participate in the study.
None showed any onco-hematological diseases, hospitalizations and
surgical procedures recent, blood transfusions, or any visible
comorbidity during the interview and signing of the consent form.
However, the low frequency of -α3.7Kb
(4.5%) found in Manacapuru perhaps can be explained because this city
has been formed by a unique indigenous ethnicity known as "Mura", and
currently, this city has smaller racial miscegenation then others
investigated in this study.[26,27]
During the
First Rubber Cycle in Brazil, an industry that demands many workers,
Manaus received a high number of immigrants from several Latin
American, European, and African countries.[28-31]
Besides, the state of Amazonas has indigenous communities with the same
hierarchical bases of the past centuries. Thus, the neo-Brazilian
population's formation took place from social and geographic
colonization, which mixed with Spaniards, English, French, Dutch,
Portuguese, and Irish, Arab-Turkish, Italian Japanese, Scandinavian and
Jewish.[32,33]
The population's characterization
through the laboratory analysis, including its hematological data and
the serum iron and ferritin dosages, in this study, allowed to
differentiate and individualize the populations. The results of the
observations and comparisons of hematological indices among alpha
thalassemia genotypes showed subtle reductions in the normal ranges and
were statistically significant, corroborant with the literature.
A
high number of individuals with hypochromic microcytic anemia, without
iron deficiency, were diagnosed with alpha thalassemia, reinforcing the
importance of the molecular techniques used in this study. Despite the
technical improvement currently offered and the constant training of
human resources, alpha thalassemia in its heterozygous form continues
to represent diagnostic difficulties for the analyst of the
conventional clinical laboratory, as well as for hematologist, who, for
the most part, are unaware of such genetic alteration, confusing it
frequently with other microcytic and hypochromic anemias. Thus, it is
essential to increase personal training and information about these
changes in our population.
Furthermore, we believe that the main
advantage of alpha thalassemia's molecular identification is the
correct distinction from iron deficiency anemia, which avoids the
possible administration of iron and other unnecessary metals to these
patients.
This study highlights thalassemia as an important public
health problem, especially in a population not yet characterized by
this disease, and reinforces the importance of assessing its frequency.
Conclusions
The present study demonstrates the importance of alpha thalassemia diagnosis in this region.
The prevalence results of -α3.7Kb
were relatively high in the majority of cities, (exception for
Manacapuru), in which many people are unaware of their genetic anemia.
Future
molecular studies might be used to describe other genetic anemias as
the pieces of beta-thalassemia or structural hemoglobinopathies as S,
C, and D that have already proven to be highly prevalent in Brazil but
not yet fully described in the northern region of Brazil, and not
studied in this paper.
Acknowledgments
Sponsorships: Financial support was provided by grants from:
• Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM) – Protocol Number: 1094/2013-FAPEAM.
•
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)
and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
(CAPES).
• The funders had no role in study
design, data collection, and analysis, decision to publish, or
preparation of the manuscript.
References
- Sakai Y, Kobayashi S, Shibata H, Furuumi H, Endo T,
Fucharoen S, Hamano S, Acharya GP, Kawasaki T, Fukumaki Y. Molecular
analysis of alpha-thalassemia in Nepal: correlation with malaria
endemicity. J Hum Genet. 2000;45(3):127-32. https://doi.org/10.1007/s100380050198 PMid:10807536
- Sabath
DE. Molecular Diagnosis of Thalassemias and Hemoglobinopathies: An
ACLPS Critical Review. Am J Clin Pathol. 2017;148(1):6-15. https://doi.org/10.1093/ajcp/aqx047 PMid:28605432
- P.
Ponka, M.J. Koury, A.D. Sheftel. Erythropoiesis, hemoglobin synthesis,
and erythroid mitochondrial iron homeostasis. G.C. Ferreira, K.M.
Kadish, K.M. Smith, R. Guilard (Eds.), Handbook of Porphyrin Science,
World Scientific Co., Singapore (2013), pp. 41-84. https://doi.org/10.1142/9789814407755_0011
- Goh
SH1, Lee YT, Bhanu NV, Cam MC, Desper R, Martin BM, Moharram R, Gherman
RB, Miller JL. A newly discovered human globin gene. Blood. 2005 Aug
15;106(4):1466-72. Epub 2005 Apr 26. https://doi.org/10.1182/blood-2005-03-0948 PMid:15855277 PMCid:PMC1895206
- Higgs DR, Weatherall DJ. The Alpha Thalassaemias. Cell Mol Life Sci. 2009;66(7):1154-62. https://doi.org/10.1007/s00018-008-8529-9 PMid:19020805
- Kasper,
Dennis L., Anthony S. Fauci, Stephen L. Hauser, Dan L. Longo, J. Larry
Jameson, and Joseph Loscalzo. eds. Harrison's Principles of Internal
Medicine, 18e. New York, NY: McGraw-Hill; 2015.
- Spier C. Wintrobeʼs Atlas of Clinical Hematology. Am J Surg Pathol. 2008;32:1428. https://doi.org/10.1097/PAS.0b013e31816955c5
- Hardison RC. (2012) Evolution of hemoglobin and its genes. Cold Spring Harb Perspect Med. 2:a011627. https://doi.org/10.1101/cshperspect.a011627 PMid:23209182 PMCid:PMC3543078
- Li CK. New trend in the epidemiology of thalassaemia. Best Pract Res Clin Obstet Gynaecol. 2017 Feb;39:16-26. https://doi.org/10.1016/j.bpobgyn.2016.10.013 PMid:27847257
- Baysal E. Huisman TJH. Detection of common deletional α-thalassemia-2 determinants by PCR. Am J Hematol. 1994;46(3):208-13. https://doi.org/10.1002/ajh.2830460309 PMid:8192150
- Tan
AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion
multiplex polymerase chain reaction assay for a-thalassemia. Blood,
vol. 98, no. 1, pp. 250-251, 2001. https://doi.org/10.1182/blood.V98.1.250 PMid:11439976
- Rosenfeld
LG, Malta DC, Szwarcwald CL, et al. Reference values for blood count
laboratory tests in the Brazilian adult population, National Health
Survey. Valores de referência para exames laboratoriais de hemograma da
população adulta brasileira: Pesquisa Nacional de Saúde. Rev Bras
Epidemiol. 2019; 22 Suppl:E190003.SUPL.2. https://doi.org/10.1590/1980-549720190003.supl.2 PMid:31596374
- GOOGLE, INC. Google Maps. Available in: http://code.google.com/apis/maps/documentation/directions/ Accessed on: March 2020.
- Higgs
DR, Goodbourn SE, Lamb J, Clegg JB, Weatherall DJ, Proudfoot NJ.
Alpha-Thalassemia caused by a polyadenylation signal mutation. Nature.
1983 Nov 24-30;306(5941):398-400. https://doi.org/10.1038/306398a0 PMid:6646217
- Cürük
MA1, Kilinç Y, Evrüke C, Ozgünen FT, Aksoy K, Yüreğir GT. Prenatal
diagnosis of Hb H disease caused by a homozygosity for the α2 polyA
mutation. Hemoglobin. 2001 May;25(2):255-8. https://doi.org/10.1081/HEM-100104034 PMid:11480787
- Karen
DF. Clinical evaluation of hemoglobinopathies: Part I. Thalassemia. The
Warde Medical Laboratory Article Archives. 2003, Volume 14, Number 2.
- Anselmo
FC, Ferreira NS, da Mota AJ, et al. Deletional Alpha-Thalassemia
Alleles in Amazon Blood Donors. Adv Hematol. 2020;2020:4170259. https://doi.org/10.1155/2020/4170259 PMid:32351571 PMCid:PMC7178540
- Foglietta
E, Deidda G, Graziani B, Modiano G, Bianco I. Detection of alpha-globin
gene disorders by a simple PCR methodology. Haematologica. 1996
Sep-Oct;81(5):387-96. Erratum in: Haematologica 1996 Nov-Dec;81(6):XVI.
PMID: 8952150.
- Wagner SC, Silvestri MC,
Bittar CM, Friedrisch JR, Silla LMR, Para C. Prevalence of thalassemias
and variant hemoglobins in patients with nonferropenic anemia. bras
hematol hemoter. 2005;27(1):37-42. https://doi.org/10.1590/S1516-84842005000100010
- WHO. Thalassaemia and other haemoglobinopathies. 2006. http://apps.who.int/gb/archive/pdf_files/EB118/B118_5-en.pdf (Acesso em 20/01/2019).
- de
Medeiros Alcoforado GH, Bezerra CM, Araújo Moura Lemos TM, et al.
Prevalence of α-thalassemia 3.7 kb deletion in the adult population of
Rio Grande do Norte, Brazil. Genet Mol Biol. 2012;35(3):594-598. https://doi.org/10.1590/S1415-47572012005000049 PMid:23055797 PMCid:PMC3459408
- de
Souza RA, Carlos AM, de Souza BM, Rodrigues CV, Pereira Gde A,
Moraes-Souza H. Α-Thalassemia: Genotypic Profile Associated with
Ethnicity and Hematological Differentiation of Iron Deficiency Anemia
in the Region of Uberaba, Minas Gerais, Brazil. Hemoglobin.
2015;39(4):264-9. https://doi.org/10.3109/03630269.2015.1037890 PMid:26182338
- Sankar
VH, Arya V, Tewari D, Gupta UR, Pradhan M, Agarwal S. Genotyping of
alpha-thalassemia in microcytic hypochromic anemia patients from North
India. J Appl Genet. 2006;47(4):391-5. https://doi.org/10.1007/BF03194650 PMid:17132905
- Lois
R. Manning, J. Eric Russell, Julio C. Padovan, Brian T. Chait, Anthony
Popowicz, Robert S. Manning, and James M. Manning. Human embryonic,
fetal, and adult hemoglobins have different subunit interface
strengths. Correlation with lifespan in the red cell. Protein Sci. 2007
Aug; 16(8): 1641-1658. https://doi.org/10.1110/ps.072891007 PMid:17656582 PMCid:PMC2203358
- Karamzade
A, Mirzapour H, Hoseinzade M, Asadi S, Gholamrezapour T, Tavakoli P,
Salehi M. α-Globin Gene Mutations in Isfahan Province, Iran. Int. J.
Hemoglobin. Res. 2014;38(3). https://doi.org/10.3109/03630269.2014.893531 PMid:24826792
- RUIS, Josué Ferreira. Manacapuru e sua história. Manacapuru: Shirley Pinheiro, 2000.
- IBGE, Biblioteca. Manacapuru Amazonas-AM: Histórico. Disponível em:<http://biblioteca.ibge.gov.br/visualizacao/dtbs/amazonas/manacapuru.pdf >. Acesso em: 29 de junho de 2020.
- Amaz RV. Revista Veredas Amazônicas - Nov - no 01, vol i, 2011. issn: 2237- 4043. 2011;I(V).
- Jakob AAE. International migration in the Brazilian Amazon. Toledo Vol. 15, Ed. 3, (2011): 422-442.
- Xavier FCC. Migrações Internacionais na Amazônia Brasileira: Impactos na Política Migratória e na Política Externa. 2012. http://repositorio.unb.br/bitstream/10482/10739/1/2012_Fernando%20Cesar%20Costa%20Xavier.pdf
- Jakob
AAE. The recent international migration in the Brazilian Amazon. REMHU,
Rev. Interdiscip. Mobil. Hum. vol.23 no.45 Brasília July/Dec. 2015. https://doi.org/10.1590/1980-8585250319880004513
- Osório, Rafael Guerreiro. O Sistema Classificatório de Cor ou Raça do IBGE. Brasília, nov.2003. Disponível em: http://www.ipea.gov.br/portal/index.php?option=com_content&view=article&id=4212 Acesso em março 2019
- Petruccelli, José Luís & Saboia, Ana Lucia(org.). Características Étnico-Raciais da População. IBGE, 2013.
Supplementary data
|
Supplementary Table 1.
Hematologic parameters characterization and levels of serum ferritin
and serum iron among alpha thalassemia 3.7kb deletion in metropolitan region of manaus.
|
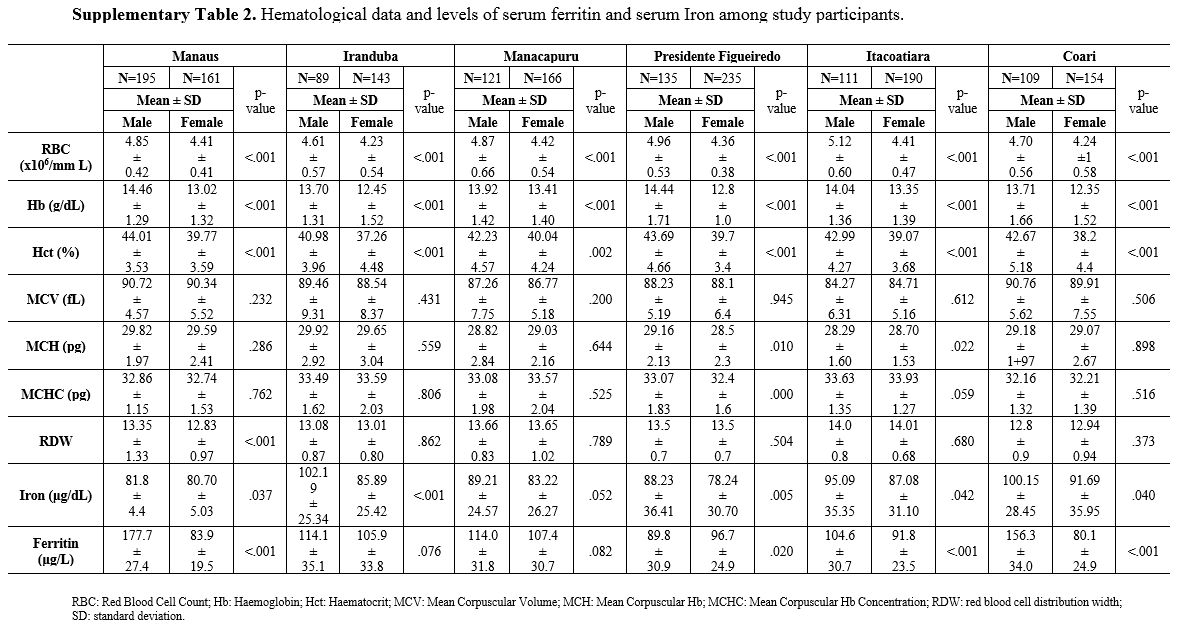 |
Supplementary Table 2. Hematological data and levels of serum ferritin and serum Iron among study participants.
|
[TOP]