Jitlada Chinsuwan1, Phatchanat Klaihmon2, Praguywan Kadegasem1, Ampaiwan Chuansumrit1, Anucha Soisamrong3, Kovit Pattanapanyasat2, Pakawan Wongwerawattanakoon4 and Nongnuch Sirachainan1*.
1 Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand.
2
Center of Excellence for Flow Cytometry, Department of Research and
Development, Faculty of Medicine Siriraj Hospital, Mahidol University,
Bangkok, Thailand.
3 Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand.
4
Division of Pediatric Nursing, Nursing Department, Faculty of Medicine
Ramathibodi Hospital, Mahidol University, Bangkok, Thailand.
Correspondence to: Prof.
Nongnuch Sirachainan, MD, Department of Pediatrics, Faculty of Medicine
Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Phayathai,
Rajathewi district, Bangkok 10400, Thailand. Tel: +66 2 201 1749; Fax:
+66 2 201 1748. E-mail:
nongnuch.sir@mahidol.ac.th
Published: November 1, 2020
Received: June 8, 2020
Accepted: October 3, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020071 DOI
10.4084/MJHID.2020.071
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Therapy
for hemophilia has evolved in the last 40 years from plasma-based
concentrates to recombinant proteins and, more recently, to non-factor
therapeutics. Along this same timeline, research in adeno-associated
viral (AAV) based gene therapy vectors has provided the framework for
early phase clinical trials initially for hemophilia B (HB) and now for
hemophilia A. Successive lessons learned from early HB trials have
paved the way for current advanced phase trials. Nevertheless,
questions linger regarding 1) the optimal balance of vector dose to
transgene expression, 2) amount and durability of transgene expression
required, and 3) long-term safety. Some trials have demonstrated unique
findings not seen previously regarding transient elevation of liver
enzymes, immunogenicity of the vector capsid, and loss of transgene
expression. This review will provide an update on the clinical AAV gene
therapy trials in hemophilia and address the questions above. A
thoughtful and rationally approached expansion of gene therapy to the
clinics would certainly be a welcome addition to the arsenal of options
for hemophilia therapy. Further, the global impact of gene therapy
could be vastly improved by expanding eligibility to different patient
populations and to developing nations. With the advances made to date,
it is possible to envision a shift from the early goal of simply
increasing life expectancy to a significant improvement in quality of
life by reduction in spontaneous bleeding episodes and disease
complications.
|
To the editor
Thromboembolism
(TE) is one of the complications of thalassemia disease. The incidences
have been reported about 0.9-4.0% in TDT and 3.9-29.0% in NTDT.[1-3]
TE's etiologies in thalassemia are an abnormal expression of PS,
platelet and endothelial activations, decreased nitric oxide, and
splenectomy.[4] In addition, MPs from red blood cells
(RBCs), platelets, endothelium, and leucocytes increase in thalassemia
diseases and play a role in TE development.[5-7] The
exposure of PS in thalassemia may contribute to the occurrence of APAs.
For example, β2GPI, a glycoprotein in circulation, when attaching to
PS, undergoes a structural change that could induce antibody formation.[8]
The prevalence of APAs in thalassemia has been reported mostly in β
thalassemia patients with the incidence rates of 42.7% of aCL-IgG,[9] 16.0% of lupus anticoagulant (LA), 30.0% of aCL-IgM, and 6.0% of aCL-IgG.[10]
Currently, there are limited studies on APAs in NTDT. In addition, the
etiology of APAs in thalassemia is still unknown. We report the
positive rates of APAs in TDT and NTDT children and demonstrate the
association of APAs with RBC, platelet, endothelial, and leucocyte MPs.Patients
with thalassemia disease and healthy controls who had normal hemoglobin
(Hb) and Hb typing were enrolled. After obtaining written consent
from parents and patients, blood was drawn for APAs – including LA
(Dade Behring Siemens Healthcare GmbH, Germany), aCL and aβ2GPI
antibodies (both IgG and IgM) (EUROIMMUN Medizinische Labordiagnostika
AG, Germany), and MPs of RBC, platelet, leukocyte, and endothelium by
flow cytometry.6 The cut-off levels were defined as >99th percentile
for aCL-IgM, aCL-IgG, aβ2GPI-IgM, and aβ2GPI-IgG. Positive APA was
determined by the subjects having at least one positive test result of
one of the APA types according to the Sydney criteria.[11] A
total of 161 subjects were divided into three groups: 55 subjects with
TDT, 44 subjects with NTDT, and 62 controls. TDT subjects had received
regular RBC transfusions (every 3-4 weeks) to maintain a
pre-transfusion mean±SD of Hb at 9.0±1.2 g/dL. After receiving RBC
transfusion, their mean Hb level was 12.3 g/dL. They required mean±SD
RBC transfusions of around 145.0±49.3 ml/kg/year. NTDT group who
required only occasional RBC transfusion of around 4.0±11.7 ml/kg/year.
As a result of regular RBC transfusion, the Hb levels in TDT and NTDT
subjects in the study were similar, with higher mean corpuscular volume
present in TDT than in NTDT subjects (Table 1).
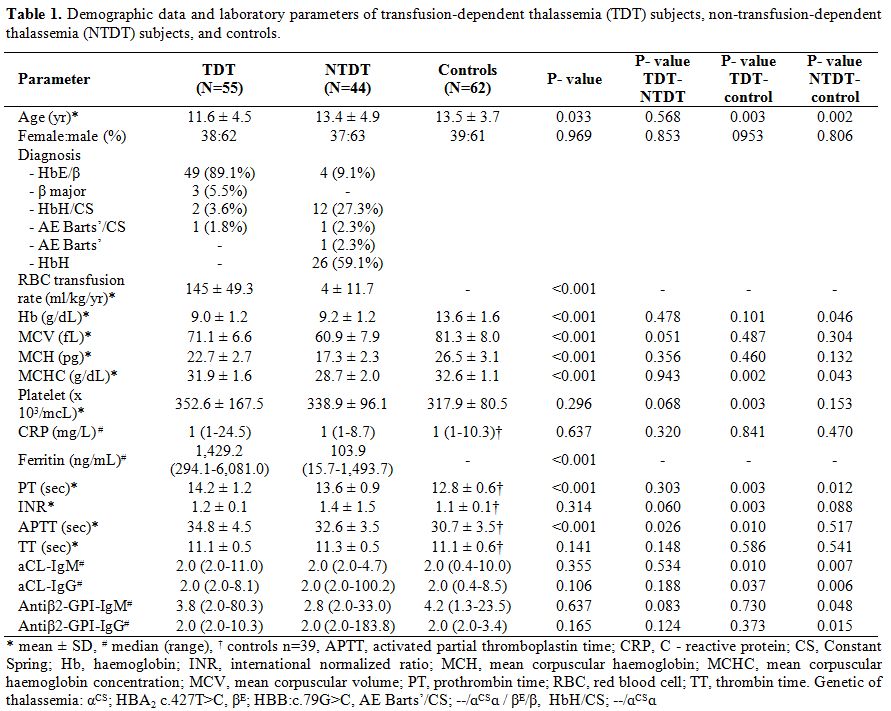 |
Table 1. Demographic
data and laboratory parameters of transfusion-dependent thalassemia
(TDT) subjects, non-transfusion-dependent thalassemia (NTDT) subjects,
and controls.
|
The
positive APA rate in all thalassemia patients as a group (23.0%) was
higher than in controls (17.9%). The positive APA rate was highest in
NTDT subjects (29.5%), and similar levels were shown between TDT
(18.2%) and control groups (17.9%), although no significant differences
were demonstrated. The LA test was positive in 14.5% of TDT subjects,
20.5% of NTDT subjects, and 12.8% of controls. When using the level
cut-off of the 99th percentile in controls to determine the positivity
of aCL (IgM = 9.99 U/mL; IgG = 8.46 U/mL) and aβ2-GPI (IgM = 23.45
U/mL, IgG = 3.44 U/mL) respectively, the aCL-IgM test was positive in
1.8% of TDT subjects and 1.6% of controls. The aCL-IgG test was
positive in 4.5% of NTDT subjects and 3.2% of controls. The aβ2GPI-IgM
and aβ2GPI-IgG were positive in 1.8% and 5.4% respectively in TDT
subjects, 4.5%, and 11.4% in NTDT subjects, and 1.6% for both IgM and
IgG in controls. The prolonged activated partial thromboplastin time
(APTT) and prothrombin time (PT) values in thalassemia subjects, when
compared to the values in controls, can be attributed to the patients
with positive APAs present in the thalassemia groups, as the APTT and
PT values were higher in thalassemia patients who had positive APA when
compared to negative APA. It is noted that a significant difference was
demonstrated only in PT values (34.1±4.2 vs. 30.9±3.7 sec, P=0.57,
14.2±1.2 vs. 12.8±0.7 sec, P=0.003 respectively).Percentages
of RBC, platelet, endothelial, and leucocyte MPs in the TDT and NTDT
groups were significantly higher than those in the controls (Table 2).
There were no significant differences in MPs percentages between the
TDT and NTDT groups except for platelet MPs, which were significantly
higher in TDT subjects than NTDT subjects (Table 2).
Aβ2GPI-IgG level significantly correlated with leucocyte (CD11b) MPs.
ACL-IgM level significantly correlated with endothelial (CD31) MPs (Table 3).
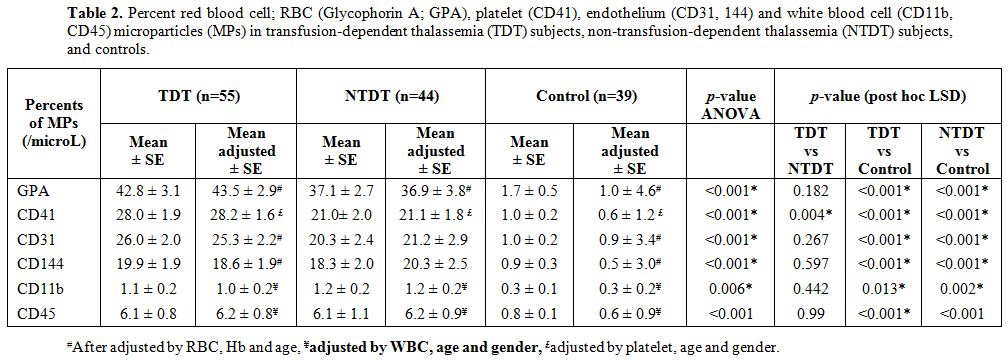 |
Table 2. Percent red blood cell; RBC
(Glycophorin A; GPA), platelet (CD41), endothelium (CD31, 144) and
white blood cell (CD11b, CD45) microparticles (MPs) in
transfusion-dependent thalassemia (TDT) subjects,
non-transfusion-dependent thalassemia (NTDT) subjects, and controls. |
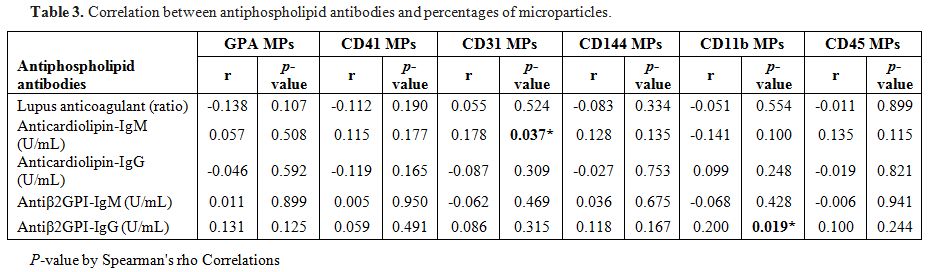 |
Table 3. Correlation between antiphospholipid antibodies and percentages of microparticles. |
Previous studies have reported a positive rate of LA in β thalassemia of 1.5-16.0%, aCL-IgG of 13.0-42.7%, and aCL-IgM of 6.0%.[9,10,12]
Our study demonstrated that subjects with positive LA in the TDT group
consisted of 14.5% and aCL-IgM of 1.8%. The differences between the
positivity rates in APAs may be due to several factors, including the
diversity of the population among the studies, the disease severity,
treatment plan (e.g., regular RBC transfusions), antibody detection
methods, and differences in cut-off value for positivity. The other
types of APAs –aβ2GPI-IgM and IgG – were also included in the present
study but did not feature any previous studies.[9,10,12]
The rates of positive aβ2GPI-IgM and IgG in the TDT group were 1.8% and
5.4%, respectively, which were higher than the aCL-IgM and IgG
positivity rates. In the NTDT group, the rates of all positive APAs
(29.5%) were higher than those in the TDT group (18.1%), although there
was no statistically significant difference demonstrated. Higher APA
positivity rates in NTDT subjects were also found for individual
antibodies, including LA, aCL-IgG, aβ2GPI-IgM, and IgG antibodies. To
our knowledge, there have been very few studies that have reported on
positive APAs in NTDT subjects, particularly in children.MPs
were higher in the TDT and NTDT groups when compared to those levels in
controls. All MPs levels (except for platelet MPs) between TDT and NTDT
groups were not significantly different. The similar MPs levels in TDT
subjects, despite more severe symptoms, may be related to the regular
RBC transfusions received by TDT subjects, and that can reduce the
amount of abnormal PS surfaces exposed. This hypothesis is supported by
a study by Atichartakarn et al.[13] The study
enrolled severe splenectomized thalassemia with pulmonary hypertension
subjects. After receiving RBC transfusions, the amount of PS surface
exposing RBC was reduced in those subjects. The report suggested that
the reduction of erythropoiesis and PS exposing cells' dilution was due
to RBC transfusion.[13] In addition, platelet activation was reduced after regular RBC transfusion. Our
study also demonstrated statistically significant correlations between
aβ2GPI-IgG and leucocyte (CD11b) MPs and aCL-IgM level and endothelial
(CD31) MPs, although the correlations were not strong. These findings
point to the likelihood that APAs in thalassemia subjects may be
related to PS's expression. The sites of PS expressed surfaces are
where glycoproteins, such as β2GPI, bind to anionic PS surfaces. After
binding to PS surfaces, β2GPI changes the conformation of the molecule
and induces antibody formation.[8] Even stronger
correlations may be demonstrated in older subject age groups because
the antibody formation may require time after exposure to the PS
expressed apoptotic cells.[14] In this study, the
lower positive APA rate in the TDT group compared to the NTDT group may
be related to the regular RBC transfusion, which may reduce the
exposure to PS expressed MPs, especially early after transfusion. In
addition to regular RBC transfusions, deferiprone has been reported to
improve immunological response, possibly from the iron chelator's
direct action and the reduction of free iron radicles.[15]
All TDT patients in the present study received iron chelation, which
was started when serum ferritin levels reached more than 1,000 ng/mL.
Deferiprone was the most prescribed medication in the present study,
accounting for 76.4% of TDT subjects. The strengths of the present
study were that it demonstrated a high prevalence rate of APAs,
especially in thalassemia patients who received an occasional
transfusion, and to our knowledge, the correlations of APAs to MPs was
first demonstrated. In
summary, high APA positive rates, associated with high MPs, were
demonstrated in a pediatric population with thalassemia disease,
especially NTDT. This suggests that MPs may play a role in APA
development. Further larger cohort and basic research studies are
required to confirm these results, better understand the occurrence of
APAs in this population, and demonstrate the risk of TE-linked APA
presence in thalassemia subjects.
Acknowledgments
The
authors would like to thank all physicians and paramedical personnel
who have been involved in treating these patients. JC performed the
research and wrote the manuscript, PK, AS and KP performed laboratory
study, AC and PW took care of the patients, and NS designed the study,
took care of the patients, and wrote the manuscript. NS is a recipient
of the Career Development Award from the Faculty of Medicine
Ramathibodi Hospital, Mahidol University, Bangkok. This study was
supported by a Ramathibodi Hospital Research Grant.
References
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833-44. https://doi.org/10.3324/haematol.2012.066845 PMid: 23729725
- Logothetis
J CM, Economidou J, Stefanis C, Hakas P, Augoustaki O, Sofroniadou K,
et al. Thalassemia major (homozygous beta-thalassemia). A survey of 138
cases with emphasis on neurologic and muscular aspects. Neurology.
1972;22(3):294-304. https://doi.org/10.1212/wnl.22.3.294 PMid: 5062264
- Cappellini
MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous
thromboembolism and hypercoagulability in splenectomized patients with
thalassaemia intermedia. Br J Haematol. 2000;111(2):467-473. https://doi.org/10.1046/j.1365-2141.2000.02376.x PMid: 11122086
- Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36-43. https://doi.org/10.1182/blood.v99.1.36 PMid: 11756150
- Kheansaard
W, Phongpao K, Paiboonsukwong K, Pattanapanyasat K, Chaichompoo P,
Svasti S. Microparticles from β-thalassaemia/HbE patients induce
endothelial cell dysfunction. Sci Rep. 2018;8(1):13033. https://doi.org/10.1038/s41598-018-31386-6 PMid: 30158562 PMCid: PMC6115342
- Klaihmon
P, Vimonpatranon S, Noulsri E, Lertthammakiat S, Anurathapan U,
Sirachainan N et al. Normalized levels of red blood cells expressing
phosphatidylserine, their microparticles, and activated platelets in
young patients with β-thalassemia following bone marrow
transplantation. Ann Hematol. 2017;96(10):1741-1747. https://doi.org/10.1007/s00277-017-3070-2 PMid: 28748286
- Youssry
I, Soliman N, Ghamrawy M, Samy RM, Nasr A, Abdel Mohsen M. Circulating
microparticles and the risk of thromboembolic events in Egyptian beta
thalassemia patients. Ann Hematol. 2017;96:597-603. https://doi.org/10.1007/s00277-017-2925-x PMid: 28168351
- Urbanus
RT, Derksen RH, de Groot PG. Current insight into diagnostics and
pathophysiology of the antiphospholipid syndrome. Blood Rev.
2008;22(2):93-105. https://doi.org/10.1016/j.blre.2007.09.001 PMid: 17964017
- Kashef
S, Karimi M, Amirghofran Z, Ayatollahi M, Pasalar M, Ghaedian MM, et
al. Antiphospholipid antibodies and hepatitis C virus infection in
Iranian thalassemia major patients. Int J Lab Hematol.
2008;30(1):11-16. https://doi.org/10.1111/j.1751-553X.2007.00916.x PMid: 18190462
- Sharma
S, Raina V, Chandra J, Narayan S. Lupus anticoagulant and
anticardiolipin antibodies in polytransfused betathalassemia major.
Hematology. 2006;11(4):287-290. https://doi.org/10.1080/10245330600954130 PMid: 17178669
- Versteeg
HH, Ruf W. Thiol pathways in the regulation of tissue factor
pro-thrombotic activity. Curr Opin Hematol. 2019;119(6):860-870. https://doi.org/10.1055/s-0039-1681102 PMid: 30861549
- Giordano
P, Galli M, Del Vecchio GC, Altomare M, Norbis F, Ruggeri L. Lupus
anticoagulant, anticardiolipin antibodies and hepatitis C virus
infection in thalassaemia. Br J Haematol. 1998;102(4):903-906. https://doi.org/10.1046/j.1365-2141.1998.00853.x PMid: 9734637
- Atichartakarn
V, Chuncharunee S, Chandanamattha P, Likittanasombat K, Aryrachai
K. Correction of hypercoagulability and amelioration of pulmonary
arterial hypertension by chronic blood transfusion in an asplenic
hemoglobin E/β-thalassemia patient. Blood. 2004;103(7):2844-2846. https://doi.org/10.1182/blood-2003-09-3094 PMid: 14645000
- Pittoni V, Isenberg D. Apoptosis and antiphospholipid antibodies. Semin Arthritis Rheum. 1998;28(3):163-178. https://doi.org/10.1016/s0049-0172(98)80033-4 PMid: 9872477
- Del
Vecchio GC, Schettini F, Piacente L, De Santis A, Giordano P, De Mattia
D. Effects of deferiprone on immune status and cytokine pattern in
thalassaemia major. Acta Haematol. 2002;108(3):144-149. https://doi.org/10.1159/000064705 PMid: 12373086
[TOP]