This study has included 102 pregnant women after having taken 90 iron tablets during antenatal visits in the Primary Health Care (PHC) facilities in Kupang, Eastern Indonesia, as published earlier.[6]
In brief, a regular complete blood count examination was conducted, and after consent, the left-over blood was kept in the fridge at +4C for further Hb analysis and DNA isolation. The erythrocyte indices calculated in this study were Mentzer Index (MI), Srivastava Index (SI), Shine & Lal Index (SLI), and England & Fraser Index (EF), the most used indices in western Indonesia. The concordance analysis was performed, and Cohen’s kappa among the indices was calculated with MI as a reference. Only those with anemia and/or MCV<80 fl and/or MCH <27pg were further examined for Hb analysis (Variant II Hemoglobin Testing System, Bio-Rad Laboratories Inc., Hercules, CA, USA) by flying the samples over to Jakarta, the capital city of Indonesia, that took 4 hours flight. The cut-off value of HbA2 <3.5% was designated as iron deficiency or possible putative α-thalassemia carriers. DNA examination was conducted to confirm the genetic mutation, if any, by using α-Globin StripAssay® (Viennalab diagnostic, cat no. 4-160) and β-Globin StripAssay® SEA (Viennalab diagnostic, cat no. 3-160), specific to Southeast Asian population. The study protocol had been reviewed and granted by the Ethical Committee Faculty of Medicine Universitas Padjadjaran no. 71/UN6.KEP/EC/2019.
The result showed that anemia (Hb <10.5 g/dL) was found in 34.3% (n=35) pregnant women, even after three months of iron supplementation (Table 1). Srivastava index (SI) was in perfect agreement or concordance (kappa = 1) to Mentzer index (MI), and other indices were poor. Interestingly, when new cut-offs by Kumar et al. were applied, the concordance of various indices to MI was increased to a substantial agreement (kappa > 0.7) (Table 1). Therefore, we further analyzed the conventional MI and modified SLI with cut-offs 13 and 891.1, respectively, and confirmed these indices with HbA2 and DNA results (Table 2). Samples with a very high HbA2 >8% were all confirmed to have G>A mutation at codon 26, known as HbE variant; therefore, Hb E carriers would have been picked up by high-performance liquid chromatography (HPLC) alone and would thus not require DNA analysis.[1] Of note, both SLI and MI did not cover HbE, leading to a possible false-negative result of some cases during screening (Table 2). Interestingly, other samples with HbA2 value >3.5% were confirmed to have no mutation in common β-globin from South East Asia (SEA) region. This negative result of SEA β-thalassemia in samples from eastern Indonesia with HbA2 >3.5% is of great interest. The population in eastern Indonesia has a different spectrum of mutation than western Indonesia, where IVS1nt-5 mutation is prevalent.[1] These samples need to be further explored for β globin gene, including its promoter region to polyA region, using the sequencing method. Alternatively, another set for β-globin examination specific to the Mediterranean area (MEA) or the India & Middle-East area (IME) might give more information.
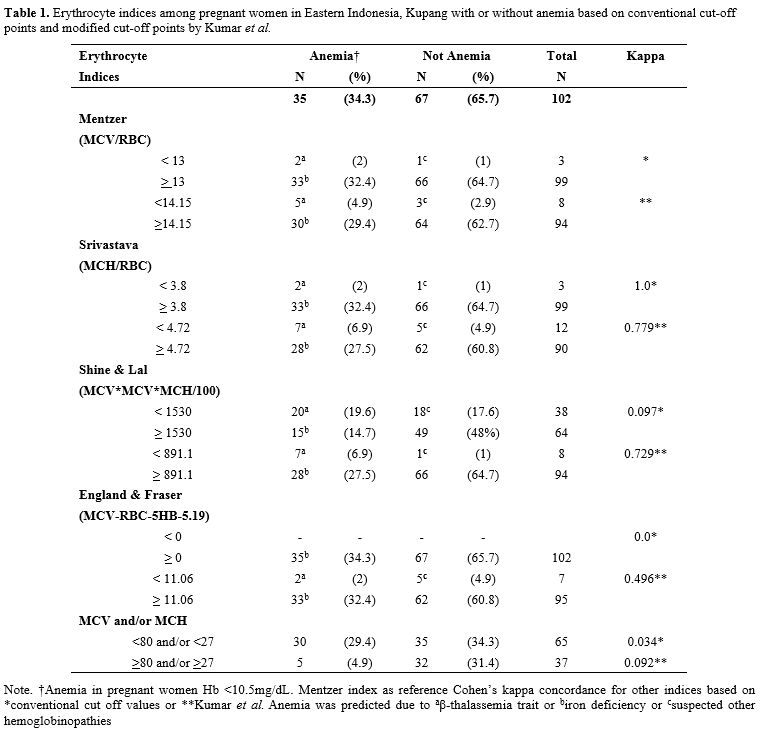 |
Table 1. Erythrocyte indices among pregnant women in Eastern Indonesia, Kupang with or without anemia based on conventional cut-off points and modified cut-off points by Kumar et al. |
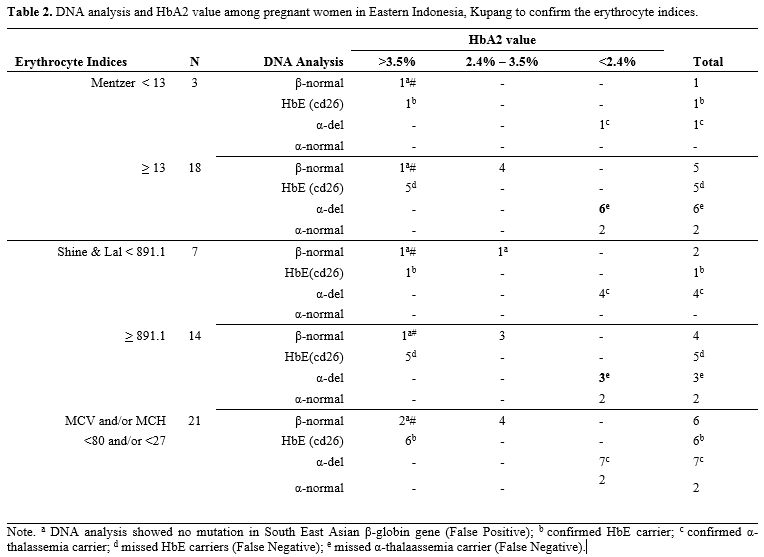 |
Table 2. DNA analysis and HbA2 value among pregnant women in Eastern Indonesia, Kupang to confirm the erythrocyte indices. |
For HbA2 <3.5%, associations could have potentially been misattributed to iron deficiency. Therefore, 12 weeks of iron supplementation needs to be given before repeated HbA2 examination. When HbA2 is still below 3.5% after iron supplementation, testing for α, δ‐ and other globin gene abnormalities should be pursued. In our study, for example, a subset of 17.5% cases with a very low HbA2 (<2.4%) was examined for α-mutation or deletion, resulting in various deletions of α-globin gen which were -α3.7del homozygous, -α3.7del heterozygous, -α4.2del heterozygous, combined -α3.7del and -α4.2del, and some had no alpha mutations. Of note, there could be a co-inheritance of α- and β-thalassemia carrier. Mutations in α- and β-globin genes may lead to a reduced or abolished globin-chain synthesis or cause structurally abnormal hemoglobin.[7] Again, both SLI and MI did not cover all samples with α-globin deletions (Table 2).
We have faced some limitations in this study. First, the blood smear examination has not been performed. Thus sickle cell disease cannot be ruled out. We acknowledge that the sample number is very small to arrive at a meaningful conclusion. However, with only limited cases, our study shows that both conventional MI and modified SLI have failed to confirm most of the HbE and α-carriers prevalent in this population, leading to false-negative results when using existing erythrocyte indices only. In this scarce resource area, assessment of MCV<80fL and MCH<27pg as the first level of detection during thalassemia screening seems to be the best way and recommended. Another limitation is that the phase of pregnancy in which the data have been collected is not ideally conducted, and the presented study is not a model for a prevention program. This study has recruited pregnant women in the third trimester, and this may raise some ethical issues as this would be too late for any prenatal intervention if required. Of note, any preventive intervention in the third trimester of pregnancy could be ethically questionable and may be unacceptable. An integrative approach for genetic counseling is needed to convey the carriership result given during antenatal screening. It is worthy to note that pregnant women might have different cut-off values in blood parameters; however, thalassemia carrier screening during antenatal care is possible.[8]
In any case, a structured prevention program is a must in an area where thalassemia carriers are prevalent. The most cost-effective and practical approach might be to screen the immediate family members for carrier identification;[9] however, some reluctance might occur in the extended family members to screen the carrier ship of their own.[10] Moreover, prevention in any groups is encouraged to decrease the thalassemia cases further, preferably in a premarital group,[11] in high school,[12] or alternatively during antenatal care in the first trimester of the first pregnancy as recommended by WHO.[13]
Therefore, it is highly recommended to deploy community screening programs and provide prospective carriers with genetic counseling to make an informed choice correctly and completely. The implementation of a simple and reliable diagnostic tool is thus essential, knowing that regarding the difficulty of prevention costs, it is commonly recognized that cure costs for thalassemia care are vastly superior.[14] The main challenge is how to do mass screening in the limited resource area, where only complete blood count examination is available.
To conclude, the erythrocyte indices conventional Mentzer index and modified Shine & Lal Index have missed most HbE carriers and α-thalassemia carriers in our study. On the contrary, simple MCV<80fl and MCH<27pg covered those carriers. Indeed, those erythrocyte indices and formulas do not intend to detect Hb E or α-thalassemia carriers. In places where the distribution of α- or β-thalassemia is unknown, mass carrier screening cannot be solely conducted by conventional erythrocyte indices. HbA2 analysis and DNA examination are needed to confirm the carriership further and are of great interest to explore the genetic variants in a particular area. Updating the epidemiological survey of hemoglobinopathies at regular intervals is thus necessary to develop effective prevention and control strategies.