Gianfranco Catalano1,2,3, Pasquale Niscola3, Cristina Banella1,2, Daniela Diverio4, Malgorzata Monika Trawinska, Stefano Fratoni5, Rita Iazzoni6, Paolo De Fabritiis1,3, Elisabetta Abruzzese3* and Nelida Ines Noguera1,2*.
1 Department of Biomedicine and Prevention, Tor Vergata University of Rome, 00133 Rome, Italy.
2 Neuro Oncohematology Unit, Santa Lucia Foundation, IRCCS. Rome, Italy.
3 Hematology Unit, Sant’ Eugenio Hospital, Tor Vergata University of Rome, Rome, Italy.
4
Hematology, Department of Precision and Translational Medicine,
Policlinico Umberto I, “Sapienza” University of Rome, Rome, Italy.
5 Department of Pathology (UOSD Anatomia Patologica) A.S.L. Roma2, Sant’ Eugenio Hospital, Rome, Italy.
6 Department of Clinical Pathology (U.O.C. Laboratorio) A.S.L. Roma2, Sant’ Eugenio Hospital, Rome, Italy.
Correspondence to: Nelida Ines Noguera. Dept. of Biomedicine and
Prevention, Tor Vergata University, 00133 Rome, Italy. Tel:
+3906501703214, Fax: +3906501703318. E-mail:
n.noguera@hsantalucia.it.
Elisabetta
Abruzzese. Hematology Unit, Saint’ Eugenio Hospital, Tor Vergata
University of Rome, 00133 Rome, Italy. E-mail: elisabetta.
abruzzese@uniroma2.it
Published: November 1, 2020
Received: August 26, 2020
Accepted: October 22, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020083 DOI
10.4084/MJHID.2020.083
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Breakpoint
cluster region - Abelson (BCR-ABL1) chimeric protein and mutated
Nucleophosmin (NPM1) are often present in hematological cancers, but
they rarely coexist in the same disease. Both anomalies are considered
founder mutations that inhibit differentiation and apoptosis, but
BCR-ABL1 could act as a secondary mutation conferring a proliferative
advantage to a pre-neoplastic clone. The 2016 World Health Organization
(WHO) classification lists the provisional acute myeloid leukemia (AML)
with BCR-ABL1, which must be diagnosed differentially from the rare
blast phase (BP) onset of chronic myeloid leukemia (CML), mainly
because of the different therapeutic approach in the use of tyrosine
kinase inhibitors (TKI). Here we review the BCR/ABL1 plus NPMc+
published cases since 1975 and describe a case from our institution in
order to discuss the clinical and molecular features of this rare
combination, and report the latest acquisition about an occurrence that
could pertain either to the rare AML BCR-ABL1 positive or the even
rarer CML-BP with mutated NPM1 at the onset. Differential diagnosis is
based on careful analysis of genotypic and phenotypic features and
anamnestic, clinical evolution, and background data. Therapeutic
decisions must consider the broader clinical aspects, the comparatively
mild effects of TKI therapy versus the great benefit that might bring
to most of the patients, as may be incidentally demonstrated by our
case history.
|
Introduction
Mutations in the NPM1 gene are the most frequent genetic abnormalities in acute myeloid leukemia (AML) and are highly specific for de novo AML.[1] The Breakpoint cluster region - Abelson (BCR-ABL)
fusion gene is the genetic hallmark of chronic myeloid leukemia (CML)
but can also be found in approximately 30% of acute lymphoblastic
leukemia (ALL) and rarely in AML (0.3–3% of newly diagnosed cases).[2–4]
In the updated World Health Organization (WHO) classification published
in 2016, AML with BCR-ABL has been introduced as a provisional new
entity.[5,6] To the best of our knowledge, the co-occurrence of the BCR-ABL fusion gene and NPM1
mutations in de novo AML has been reported in only a few cases. In this
review, we analyzed all the BCR/ABL1 plus NPMc+ published cases since
1975 and a case from our institution to present common clinical and
molecular features of this rare disease.
t(9;22)(q34.1;q11.2) BCR-ABL.
Among human cancers, AMLs are relatively genetically simple and stable
diseases featuring the fewest mutations variety and average. AML
genomes contain a median of 13 coding mutations (single nucleotide
variants and insertion/deletions) and an average of less than one
gene-fusion event.[7,8] Most of the fusions derive
from translocation events, and Philadelphia chromosome
t(9;22)(q34.1;q11.2), generating the BCR-ABL1 chimeric protein, was the
first genetic aberration associated with human cancer: Chronic myeloid
leukemia (CML). BCR-ABL activates proliferation signaling pathways (RAS
and STAT5, STAT1 and STAT6 signaling, PI3-K and AKT/PKB pathways),
analogously to PML/RARa in APL inhibits PTEN,[9,10]
interferes with the focal adhesion complex (PAXILLIN, FAK), induces
abnormal integrin signaling (FAK/CRK-L/SDF-1) and has anti-apoptotic
activity (PI63K/Akt/STAT5). In addition, BCR-ABL has been shown to
generate a “mutator” phenotype downregulating homeostatic controls and
DNA repair pathways and promoting the expression of
DNA-polymerase-beta, which is prone to copy errors during DNA
replication.[11] Since no CML-BP with lymphoid
phenotype carrying the NPM1c+ mutation was ever reported, we will not
address the subject of Ph1+ALL. Contrarily to what could be expected
only a few years ago, myeloid neoplasms carrying the BCR-ABL
transcripts are a composite subset of hematological disorders. If the
principal disease, CML, in which BCR-ABL1 is involved, has
well-characterized features and standardized diagnosis and therapy, the
picture is composite for the other neoplastic diseases. 2016 WHO
classification of myeloid neoplasms and acute leukemia distinguish,
other than CML, two more entities: one mixed phenotype acute leukemia
(MPAL) with BCR-ABL1, and the provisional AML with BCR-ABL1.[6]
Since no CML-BP with lymphoid phenotype carrying the NPM1c+ mutation
was ever reported, we will not address the subject of Ph1+ALL.
Nucleophosmin.
Nucleophosmin (NPM1) is present in high quantities in the granular
region of nucleoli but shuttles between nucleus and cytoplasm, acting
as a chaperone. Chaperones are molecules that associate with target
proteins, organize their structure, convoy them to the appropriate
place, and molecular aggregate but are not part and have no function in
that aggregate.[12,13] NPM1 has been identified as the most frequently mutated gene in AML patients, accounting for about 30% of cases,[14–16]
the vast majority of which with normal karyotype. At onset, NPM1
mutation associates with a less severe prognosis, but clonal evolution
can lead to additional genetic abnormalities and worst prognosis.[1,16,17]
NPM1 gene mutations in AML lead to a new C-terminus sequence in the
mutant protein, that, as compared to the wild-type protein, lacks the
nucleolar binding site and acquires a nuclear export signal: mutated
NPM1 is confined to cytoplasm, its absence from the nucleus seems to be
the basis for the oncogenic phenotype since the protein plays a role in
chromatin remodeling, centrosome duplication, DNA replication,
recombination, transcription, and repair as well as in the control of
cell cycle progression and survival in response to a variety of stress
stimuli.[12,18–21]
The Paradigm of Leukemogenesis.
Mutations within a cell can influence the rate of acquisition of other
lesions. After the initiating mutation, there might be a gradual
accumulation of additional genetic alterations or accelerated
progression due to genomic instability or catastrophic genetic events,
including chromothripsis.[22–27] The number of
identifiable driver mutations differs between AML cases. Although most
cases harbor three or more identifiable drivers at the time of clinical
presentation, human sequencing data describe many AML with only one or
two identifiable driver mutations.[24,28]
According to the model of Gilliland and Griffin, the paradigm of
leukemogenesis features a class II mutation as leukemia-initiating
event, causing inhibition of differentiation and apoptosis, cooperating
with a class I mutations conferring a proliferative advantage to the
clone.[29,30] In 2013 the Cancer Genome Atlas
Research Network classified three sets of genes with the strongest
patterns of mutual exclusivity. For the purpose, they used whole genome
or whole exome sequencing and statistical analysis of 200 de novo AML
cases selected from a set of more than 400 samples to reflect a
real-world distribution of subtypes. The first set comprised the
transcription-factor fusion genes and mutations involving NPM1, RUNX1, TP53, and CEBPA, the second set the mutations in genes encoding FLT3
or other tyrosine kinases (TK), serine-threonine kinases, protein
tyrosine phosphatases, RAS family proteins, and the third set included
mutations in ASXL1 and genes encoding components of the cohesin complex, other myeloid transcription factors, and other epigenetic modifiers.[7] The association of BCR-ABL1 and mutated NPM1 in the same clone is unusual but not contradictory to either of the models if NPM1 is the founder, class II mutation, and BCR-ABL1
acts a class I mutation, conferring a proliferative advantage to the
affected cells. BCR-ABL1, even though capable of transforming
hemopoietic stem cells single-handed and causing per se CML and diverse
acute leukemias (Ph1+ ALL, MPAL and AML), could be working as a class I
mutation[31] since the molecular aberration was found in tumor subclones and even in oligoclones in otherwise normal bone marrow.[32,33]
However, would that be possible to reverse the rank of the mutations,
as in a CML blastic phase (CML-BP) clone carrying mutated NPM1 evolving
from an NPM1-negative chronic phase disease? Moreover, how to
discriminate between de novo Philadelphia positive AML and a CML
diagnosed at BP onset? Even though identical regarding two substantial
features of the genetic profile, the two conditions must have different
biology. The presence of BCR-ABL1 protein ab initio, thus in
tumor-initiating cells and all the disease clones, must confer the
phenotype, natural history, and clinic of CML. Conversely, the
emergence of a BCR-ABL positive clone as a type I mutation in an NPM1
mutation expressing clone is not more than a concomitant feature in the
characteristic of acute leukemia, in a way not entirely different from
a FLT3 activating mutation (Figure 1).
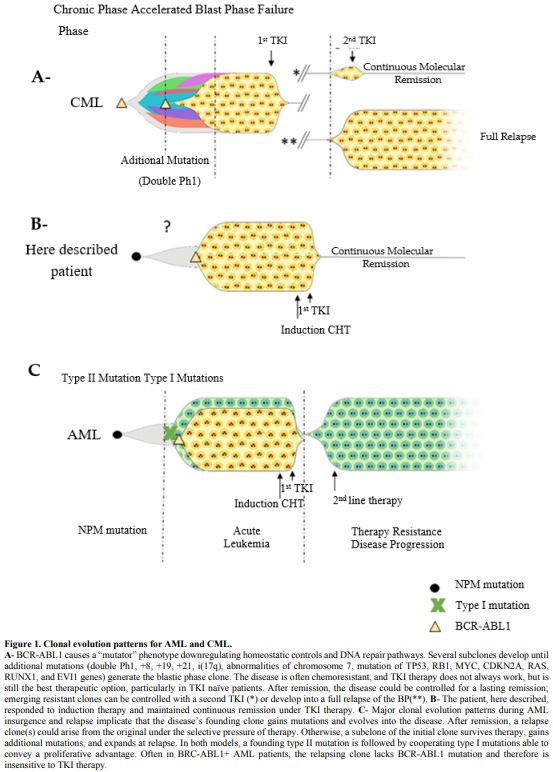 |
Figure
1. Clonal evolution patterns for AML and CML.
A-
BCR-ABL1 causes a “mutator” phenotype downregulating homeostatic
controls and DNA repair pathways. Several subclones develop until
additional mutations (double Ph1, +8, +19, +21, i(17q), abnormalities
of chromosome 7, mutation of TP53, RB1, MYC, CDKN2A, RAS, RUNX1, and
EVI1 genes) generate the blastic phase clone. The disease is often
chemoresistant, and TKI therapy does not always work, but is still the
best therapeutic option, particularly in TKI naïve patients. After
remission, the disease could be controlled for a lasting remission;
emerging resistant clones can be controlled with a second TKI (*) or
develop into a full relapse of the BP(**). B- The patient, here
described, responded to induction therapy and maintained continuous
remission under TKI therapy. C- Major clonal evolution patterns during
AML insurgence and relapse implicate that the disease’s founding clone
gains mutations and evolves into the disease. After remission, a
relapse clone(s) could arise from the original under the selective
pressure of therapy. Otherwise, a subclone of the initial clone
survives therapy, gains additional mutations, and expands at relapse.
In both models, a founding type II mutation is followed by cooperating
type I mutations able to convey a proliferative advantage. Often in
BRC-ABL1+ AML patients, the relapsing clone lacks BCR-ABL1 mutation and
therefore is insensitive to TKI therapy.
|
NPM1 Mutated and BCR-ABL1 Positive Myeloid Neoplasm
To the best of our knowledge, only a few cases of de novo AML with BCR-ABL1 and NPM1 mutations were published in the last decades (Table 1). Bacher et al. describe a case with normal karyotype AML (FAB M4) and an NPM1 mutation, the occurrence of a Philadelphia positive subclone in an NPM1
mutated AML patient emerging at relapse of the disease. This patient
received initially high dose of chemotherapy, and also intensive
chemotherapy with high dose cytarabine and mitoxantrone after relapse,
unfortunately dying from progression 26 months from diagnosis.[31] Single cases with Philadelphia positive subclones in NPM1 mutated AML had previously been reported by Suzuki et al.[34] and Verhaak et al. [35] Palmisano et al. reported a patient who maintained the NPM1 mutation at relapse of the disease, whereas the t(9;22) was lost.[36]
Konoplev et al. analyzed NPM1 and ABL1 genes, often mutated in AML and
CML-BP patients, respectively, to gather insights into the relationship
between Ph+ AML and CML-BP. They studied 9 Ph+ AML and 5 CML-BP
patients at the onset. Two out of 9 Ph+ AML patients had NPM1 mutations
and were alive 36 and 71 months after diagnosis. All Ph+ AML had no
mutation in the ABL1 sequence, no NPM1 mutations were identified in the CML-BP group, and one CML-BP patient had ABL1 mutation. The Authors argue that Ph+ AML is distinct from CML-BP.[36] Reboursiere et al. describe one case harboring a BCR-ABL p210 transcript level of approximately 10% with NPM1
gene mutation. The patient received induction therapy with
mitoxantrone, daunorubicin, and cytosine arabinoside and two courses of
high-dose cytosine arabinoside consolidation therapy followed by
allogenic hematopoietic stem cell transplantation (allo-HSCT). Eleven
years after allo-HSCT, the patient remained in continuous complete
molecular remission.[37] Mattioti et al. report a
patient diagnosed with AML harboring a complex three-way translocation
t(9;22;12)(q34;q13;q11) encoding for two isoforms of BCR-ABL transcript (b3a2;b2a2) and a concomitant type A mutation in the NPM1
gene. The patient was started on initial cytoreductive treatment with
hydroxyurea for ten days and was subsequently treated with
second-generation tyrosine kinase inhibitor (TKI) dasatinib due to the
central nervous system’s high risk involvement and extramedullary
localization. Unfortunately, the patient died after 3 months of
treatment.[38] Studies regarding CML have shown that
in some cases transcript, b2a2 has slower molecular and inferior
response rates to TKI and a poorer long-term outcome,[39]
but at present, no reliable data are available regarding the prognostic
value of the different transcripts in AML. Neuendorff et al., in an
exhaustive review, describe 6 NPM1 mutated at primary diagnosis out of 126 cases of de novo BCR-ABL1+ AML. At least 3 of these 6 NPM1 BCR-ABL1+ AML patients were long-term survivors, which notwithstanding the exiguity of cases, is a large percentage.[40]
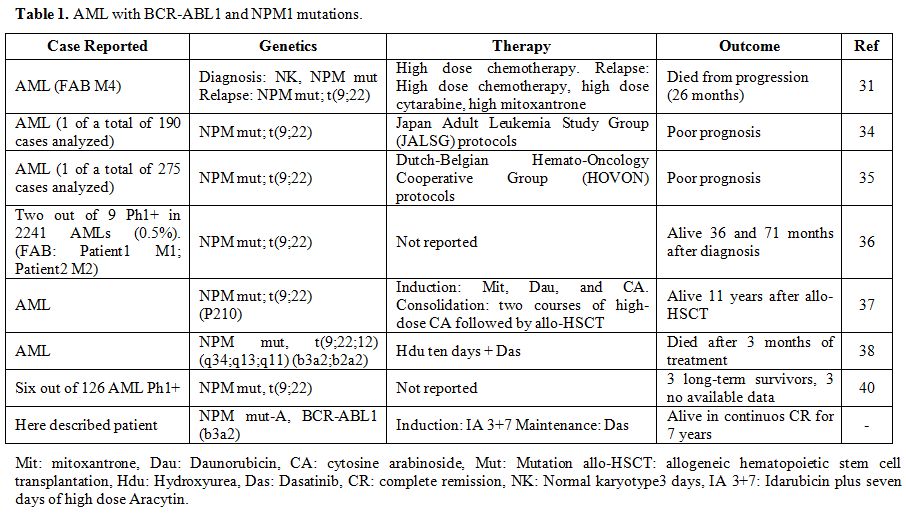 |
Table
1.AML with BCR-ABL1 and NPM1 mutations.
|
From the clinical point of view, sometimes the presence of the BCR-ABL1
hybrid transcript could not clarify the adjudication since a precise
distinction between AML and CML-BP at onset is still to be defined,
which poses problems of diagnosis and therapy, most of all about the
timing and efficacy of TKI therapy. Notwithstanding the rarity of
cases, it seems clear that AML with BCR-ABL1, in general, does not always respond well to TKI therapy ;[40] conversely, a TKI naïve CML must benefit from TKI therapy.[41]
Experience in Our Institution.
Here we want to narrate the case of a patient, 43 years of age, male,
diagnosed in April 2013 at a different country institution supposedly
with a CML onset in BP and treated with 3 days Idarubicin plus seven
days of high dose Aracytin (IA 3+7) resulting in complete hematological
remission on day 26. The patient had experienced a series of severe
complications during the induction therapy. The routine molecular
assessment had documented positivity for BCR-ABL1 (p210-B3A2)
translocation and NPM1 mutation A.[42,43]
The patient, with residual hepatic toxicity, was prescribed Dasatinib
100mgr as maintenance therapy. The diagnosis was well documented as for
the extension of the search for the mutations but essential since we
were not detailed about the FAB and immunophenotype of the blasts,
quantities of the mutated transcripts, or about any other clinical
aspect that would explain the choice of CML-BP over AML with BCR-ABL1+.
In our opinion is of interest that the case was labeled as CML-BP, yet
the clinical perspective and indication were that of a de novo AML with
BCR-ABL1: intensive induction chemotherapy with additional TKI maintenance therapy, then consolidation and allo-SCT.
On day 45, our bone marrow evaluation showed hematologic morphologic remission, molecular remission of the BCR-ABL1
transcript (p210 transcript was 0.069% with MR3 sensitivity, p190 was
0,0% negative MR3 sensitivity) and molecular remission of the NPM1 mutated transcript (0.025 of NPM1 mutation A copies every 104 copies of ABL, cut off value 0.03).[44]
Of note that only a small percentage, less than 10%, of CML-BP
patients, achieves molecular remission after frontline chemotherapy
plus TKI therapy.[41] In contrast, almost one in three of AML NPM1 mutated patients achieve molecular remission 30 days after frontline therapy.[45]
The search for a compatible donor among siblings was unsuccessful, we
consulted with the patient. We agreed to postpone intensive
chemotherapy mainly for the high risk of a recurrence of the intestinal
bleeding, at the same time the patient did not agree to start a search
for an unrelated compatible donor. One year since onset p210 transcript
was undetectable with MR5 sensitivity, we stop essaying p190, and the NPM1 mutated transcript remained in molecular remission (0.03 of NPM1 mutation A copies every 104 copies of ABL,
cut off value 0.03). After more than 6 years, the patient is still in
continuous profound molecular remission of the BCR-ABL1 (undetectable
transcript with MR5 sensitivity) and remission of the NPM1 transcripts.
Still assuming Dasatinib, the dose was interrupted for 30 days and then
reduced by half to 50 mg per day due to a chemical pleuritis about 12
months ago. The interruption and lowered dose did not cause any
variation in the molecular remission of both transcripts. Since all
disease-free survival (DFS) curves tend to plateau after 2-3 years of
follow-up and relapse after 5 years of DFS are rare events, we may say
the patient was fortunate not to undergo intensive chemotherapy and
allo-SCT.
Nevertheless, is this patient eligible for ending TKI
therapy? It all depends on the diagnosis. A CML-BP in remission is not
eligible in any case for stopping TKI therapy. Conversely, an AML in
continuous molecular remission after 6 years could be considered for
ending the treatment.
Differential Diagnosis
CML
primary blast phase is an infrequent event, and the secondary blast
phase is usually marked out by patients’ medical history. In the lack
of a previous CML diagnosis, most BP cases must carry the clinical and
morphologic stigmata of the chronic phase. Thus anamnestic and clinical
features, histology and morphology, immunophenotype, and genetic can be
of help. The presence of basophilia often accompanies blast cell
expansion and disease acceleration in CML, whereas it is not a common
feature in de novo AML, the same for splenomegaly. In BP, bone marrow
megakaryocytic count is increased in most cases with perisinusoidal
distribution and no clusters, hypolobation of nuclei, and the presence
of micromegakaryocytes. Broadly 75% of BP show a myeloid phenotype and
in more than 80% of cases feature additional genetic abnormalities
(double Ph1, +8, +19, +21, i(17q), abnormalities of chromosome 7,
mutation of TP53, RB1, MYC, CDKN2A, RAS, RUNX1, and EVI1 genes).[46]
Of note, ABL1 TK domain mutations are typical of CML-BP and are not
restricted to patients with prior TKI exposure. In a study of
unselected TKI-naïve CML-BP patients, 5 of 19 patients had ABL1 mutations.[45] ABL1 mutations have not been reported in Ph+ AML patients.[15] Under the genotypic profile, there is a link between BCR-ABL1
rearrangement and some features associated with the lymphoid phenotype.
Nacheva et al. studying 9 de novo AML Ph1+, 6 myeloid, and three
biphenotypic leukemia showed that BCR-ABL1+AML blasts often are
burdened by aberration commonly associated with lymphoid lineage tumors
(deletions of IKZF and CDKN2A/B and concomitant loss within the immunoglobulin and T cell receptor gene complexes) that are all also found in BCR-ABL1+ ALL and CML chronic phase, but not in myeloid CML-BP.[47]
The
presence of lymphoid markers, not sufficient for a classification as
MPAL according to WHO, is in line with a finding by Atfy et al., who
found in all nine cases of de novo Ph1+ AML: CD33 and CD13 markers, and
CD64 in 8 of them. MPO was positive in 9/9 patients by flow cytometry.
The B-lymphoid marker CD79a was positive in one, T-lymphoid marker CD7
in 4, CD24 in one case, and CD19 was found in two AML cases that could
be considered as FAB M2. Seven of the nine AML patients had an aberrant
expression of lymphoid markers. Stem cell markers CD34 were positive in
6/9, and TDT was positive in 1/9 cases. According to the FAB, one case
was diagnosed as M0, 3 cases as M1, 4 cases as M2, and one case as M4.[48]
In
a recent study among 46 cases of myeloid BP, 76% expressed CD34, and
74% expressed CD117. Myeloperoxidase expression was noted in a variable
proportion of precursor cells in 85% of cases. TDT was expressed in 37%
cases, 14 cases expressed markers outside of the standard myeloid
phenotype, and two expressed markers of more than one lineage (B or/and
T).[49]
In a retrospective study of 477 BP
cases in 20 years encompassing the introduction of TKI therapy, Jain P
et al. found that, for 77 patients diagnosed as BP at the onset,
first-line treatment included TKI alone (24 patients; 34%), TKI plus
chemotherapy (41 patients; 58%), non-TKI-based therapies (2 patients;
3%). Clonal evolution under therapy pressure must play a role since
patients with de novo BP had a longer overall survival time (OS)
compared with patients who transformed from CML-Chronic Phase/CML-Acute
Phase (P<.0001). The most effective treatment option was the
combination of a TKI with chemotherapy. Patients who achieved
morphologic hematologic remission (MHR) or complete cytogenetic
remission (CCyR) or major molecular response (MMR) after initial BP
treatment had a significantly longer failure-free survival (FFS)
(P<.0001) and the achievement of MHR and/or CCyR emerged as the most
significant independent predictors of survival.[41]
In a 2019 review, Soverini et al. state that 2 to 5% of CML patients
present in accelerated phase (AP) and 2 to 7% in BP and that, as a
whole, AP/BP patients display a high degree of genetic instability,
with an accumulation of additional genetic and cytogenetic
abnormalities that reduce sensitivity to TKI. However, the paper does
not address the genetics of de novo AP/BP patients.[50]
In a study published in 2015, Klco et al. confirm that among 71 patients with de novo AML, 18 patients carrying NPM1
mutated alleles were cleared below the threshold of 2,5% (5% of cells)
at day 30 from induction therapy start, and those patients have the
best chances to have a long first remission. Seemingly type I mutations
as FLT3, KRAS, or NRAS
were usually cleared on day 30, suggesting that subclones containing
these mutations may be highly sensitive to induction chemotherapy, but
of course, those patients tend to relapse early and have a poor
prognosis.[51]
Thus would not be unusual that in
our case, if considered as an AML, the molecular profile was nearly
negative for both mutations on day 45. Conversely, a CML-BP in
hematological remission after just one intensive chemotherapy cycle and
after no more than 18 days of TKI therapy should register at least a
substantial regrowth of clonal BCR-ABL1
positive hematopoiesis. As exposed before, there are several
suggestions but not certainty about discriminating de novo Ph1+AML and
CML-BP at the onset. In the absence of a previous CML history,
differential diagnosis is based on the global analysis of histologic,
immunophenotype, and genetic features, which in most cases singularly
are not decisive in differentiating the two conditions, but taken all
together may lead to a possible assignation. We summarize the
differential characteristics in Table 2.
It is interesting as some statistical modeling reports implicate the
role of functional NPM1 in conveying tumorigenic signals from the
BCR-ABL1 oncoprotein to ribosome biogenesis, affecting cellular growth.[52,53] Thus, in theory, NPM1
mutation could hamper, in part, BCR-ABL1 oncogenic phenotype, which
explains the rarity of the finding and renders NPM1 a highly improbable
candidate for BP transition.
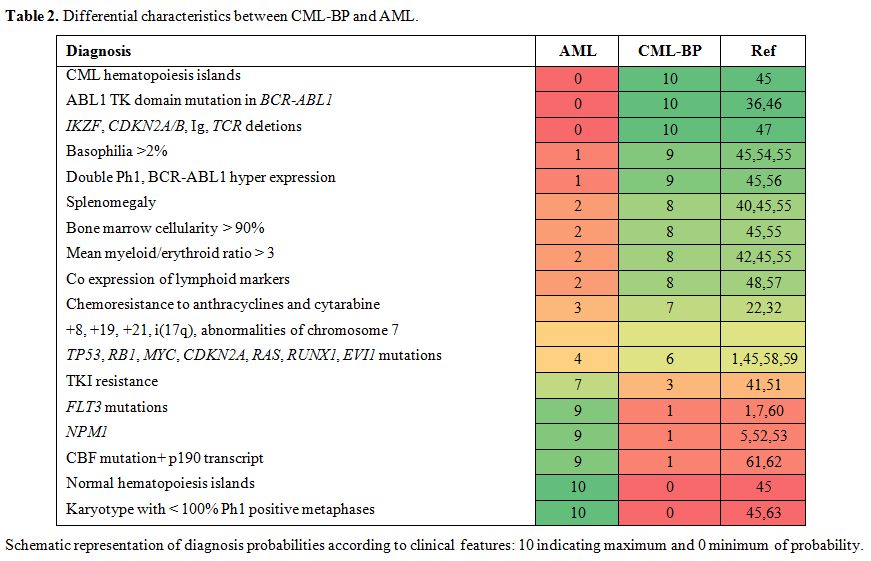 |
Table 2. Differential characteristics between CML-BP and AML.
|
Conclusions
In
conclusion, there seems to be only one clear precedent of CML-BP
carrying the NPM1 mutation, convincing under the clinical point of view
since we are given no information about the mutational status of the
CP,[64] whereas double mutated NPM1 BCR-ABL1+ AML,
although rare, has been clearly devised as part of the AML with mutated
NPM1 classification. We feel that NPM1 mutation presence has to be
considered decidedly as a sign of AML rather than BP. Therapeutic
decisions must consider the broader clinical aspects, the comparatively
mild side-effects of TKI therapy versus the great benefit that might
bring to most of the patients, as our case history may incidentally
demonstrate it. Even considering all the premises, and even after the
chemical pleuritis he suffered after five years of Dasatinib at 100 mg
per day, a complication promptly resolved with 30 days interruption,
diuretic, and corticoid therapy. However, we are not counseling to end
TKI therapy; since there are no antecedents to guide us, we rather play
safe continuing a course of action that was highly effective and with
affordable side effects so far.
References
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik
VI, Paschka P, Roberts ND et al. Genomic classification and prognosis
in acute myeloid leukemia. N Engl J Med. 2016; 374(23):2209-2221. https://doi.org/10.1056/NEJMoa1516192 PMid:27276561 PMCid:PMC4979995
- Keung
YK, Beaty M, Powell BL, Molnar I, Buss D, Pettenati M. Philadelphia
chromosome positive myelodysplastic syndrome and acute myeloid leukemia
- Retrospective study and review of literature. Leuk Research 2004;
28(6):579-86. https://doi.org/10.1016/j.leukres.2003.10.027 PMid:15120934
- Soupir
CP, Vergilio JA, Dal Cin P, Muzikansky A, Kantarjian H, Jones D et al.
Philadelphia chromosome-positive acute myeloid leukemia: A rare
aggressive leukemia with clinicopathologic features distinct from
chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol
2007; 127(4): 642-50. https://doi.org/10.1309/B4NVER1AJJ84CTUU PMid:17369142
- Cuneo
A, Ferrant A, Michaux JL, Demuynck H, Boogaerts M, Louwagie A et al.
Philadelphia chromosome-positive acute myeloid leukemia:
Cytoimmunologic and cytogenetic features. Haematologica 1996;
81(5):423-7.
- Döhner H, Estey E, Grimwade
D, Amadori S, Appelbaum FR, Büchner T et al. Diagnosis and management
of AML in adults: 2017 ELN recommendations from an international expert
panel. Blood 2017; 129(4): 424-447. https://doi.org/10.1182/blood-2016-08-733196 PMid:27895058 PMCid:PMC5291965
- Arber
DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The
2016 revision to the World Health Organization classification of
myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-405. https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Ley
TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson G et al.
Genomic and epigenomic landscapes of adult de novo acute myeloid
leukemia. N Engl J Med. 2013; 368: 2059-74. https://doi.org/10.1056/NEJMoa1301689 PMid:23634996 PMCid:PMC3767041
- Papayannidis
C, Sartor C, Marconi G, Fontana MC, Nanni J, Cristiano G et al. Acute
myeloid leukemia mutations: Therapeutic implications. Int J Mol Sci
2019. 20: (11), https://doi.org/10.3390/ijms20112721 PMid:31163594 PMCid:PMC6600275
- Noguera
NI, Piredda ML, Taulli R, Catalano G, Angelini G, Gaur G et al.
PML/RARa inhibits PTEN expression in hematopoietic cells by competing
with PU.1 transcriptional activity. Oncotarget 2016, 7: 66386-66397. https://doi.org/10.18632/oncotarget.11964 PMid:27626703 PMCid:PMC5341808
- Panuzzo
C, Crivellaro S, Carrà G, Guerrasio A, Saglio G, Morotti A. Bcr-abl
promotes pten downregulation in chronic myeloid leukemia. PLoS ONE
2014; 9: e110682. https://doi.org/10.1371/journal.pone.0110682 PMid:25343485 PMCid:PMC4208795
- Huret
J-L, Ahmad M, Arsaban M, Jacquemot-Perbal M-C, Le Berre V, Malo A et
al. Atlas of Genetics and Cytogenetics in Oncology and Haematology
Staff http://AtlasGeneticsOncology.org Atlas of Genetics and
Cytogenetics in Oncology and Haematology. Atlas Genet Cytogenet Oncol
Haematol 2015.
- Okuwaki M. The structure
and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar
acidic protein. J Biochem. 2008; 143: 441-8. https://doi.org/10.1093/jb/mvm222 PMid:18024471
- Yip
SP, Siu PM, Leung PHM, Zhao Y, Yung BYM. The multifunctional nucleolar
protein nucleophosmin/NPM/B23 and the nucleoplasmin family of proteins.
Protein Reviews 2011; 15: 213-252. https://doi.org/10.1007/978-1-4614-0514-6_10 PMCid:PMC7121557
- Brown
P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G et al. The
incidence and clinical significance of nucleophosmin mutations in
childhood AML. Blood 2007; 110:979-85. https://doi.org/10.1182/blood-2007-02-076604 PMid:17440048 PMCid:PMC1924773
- Calvo
KL, Ojeda MJ, Ammatuna E, Lavorgna S, Ottone T, Targovnik HM et al.
Detection of the nucleophosmin gene mutations in acute myelogenous
leukemia through RT-PCR and polyacrylamide gel electrophoresis. Eu J
Haematol. 2009, 82: 69-72. https://doi.org/10.1111/j.1600-0609.2008.01155.x PMid:18801061
- Falini
B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E et al. Acute
myeloid leukemia with mutated nucleophosmin (NPM1): Is it a distinct
entity? Blood 2011; 117: 1109-20. https://doi.org/10.1182/blood-2010-08-299990 PMid:21030560
- Martínez-Losada
C, Serrano-López J, Serrano-López J, Noguera NI, Garza E, Piredda L et
al. Clonal genetic evolution at relapse of favorable-risk acute myeloid
leukemia with NPM1 mutation is associated with phenotypic changes and
worse outcomes. Haematologica 2018; 103: e400-e403. https://doi.org/10.3324/haematol.2018.188433 PMid:29622659 PMCid:PMC6119134
- Federici
L, Falini B. Nucleophosmin mutations in acute myeloid leukemia: A tale
of protein unfolding and mislocalization. Protein Sci. 2013; 22:
545-56. https://doi.org/10.1002/pro.2240 PMid:23436734 PMCid:PMC3649256
- Noguera
NI, Song MS, Divona M, Catalano G, Calvo KL, García F et al.
Nucleophosmin/B26 regulates PTEN through interaction with HAUSP in
acute myeloid leukemia. Leukemia 2013; 27: 1037-43. https://doi.org/10.1038/leu.2012.314 PMid:23183427
- Colombo
E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: A
possible therapeutic target in hematological diseases. Oncogene 2011;
30:2595-609. https://doi.org/10.1038/onc.2010.646 PMid:21278791
- Colombo
E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine J-C
et al. Nucleophosmin Is Required for DNA Integrity and p19Arf Protein
Stability. Mol Cell Biol. 2005; 25(20):8874-86. https://doi.org/10.1128/MCB.25.20.8874-8886.2005 PMid:16199867 PMCid:PMC1265791
- Stephens
PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al. Massive
genomic rearrangement acquired in a single catastrophic event during
cancer development. Cell 2011; 144(1):27-40. https://doi.org/10.1016/j.cell.2010.11.055 PMid:21215367 PMCid:PMC3065307
- Fontana
MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, Ghelli Luserna Di
Rorá A et al. Chromothripsis in acute myeloid leukemia: Biological
features and impact on survival. Leukemia 2018; 32:1609-1620. https://doi.org/10.1038/s41375-018-0035-y PMid:29472722 PMCid:PMC6035145
- Welch
JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC et al. The origin
and evolution of mutations in acute myeloid leukemia. Cell 2012;
150(2):264-78. https://doi.org/10.1016/j.cell.2012.06.023 PMid:22817890 PMCid:PMC3407563
- Noguera
NI, Catalano G, Banella C, Divona M, Faraoni I, Ottone T et al. Acute
promyelocytic Leukemia: Update on the mechanisms of leukemogenesis,
resistance and on innovative treatment strategies. Cancers 2019;
11(10):1591. https://doi.org/10.3390/cancers11101591 PMid:31635329 PMCid:PMC6826966
- Krönke
J, Bullinger L, Teleanu V, Tschürtz F, Gaidzik VI, Kühn MWM et al.
Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood
2013; 122(1):100-8. https://doi.org/10.1182/blood-2013-01-479188 PMid:23704090
- Jan
M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR et al.
Clonal evolution of preleukemic hematopoietic stem cells precedes human
acute myeloid leukemia. Sci Trans Med 2012; 4(149):149ra118.
doi:10.1126/scitranslmed.3004315. https://doi.org/10.1126/scitranslmed.3004315 PMid:22932223 PMCid:PMC4045621
- Potter
N, Miraki-Moud F, Ermini L, Titley I, Vijayaraghavan G, Papaemmanuil E
et al. Single cell analysis of clonal architecture in acute myeloid
leukaemia. Leukemia 2019; 33(5):1113-1123. https://doi.org/10.1038/s41375-018-0319-2 PMid:30568172 PMCid:PMC6451634
- Gilliland
DG. Molecular genetics of human leukemias: New insights into therapy.
Sem Hematol 2002.; 39:6-11. doi:10.1053/shem.2002.36921.
https://doi.org/10.1053/shem.2002.36921 PMid:12447846
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 39:6-11. https://doi.org/10.1182/blood-2002-02-0492 PMid:12176867
- Bacher
U, Haferlach T, Alpermann T, Zenger M, Hochhaus A, Beelen DW et al.
Subclones with the t(9;22)/BCR-ABL1 rearrangement occur in AML and seem
to cooperate with distinct genetic alterations. Br J Haematol; 2011;
152(6):713-20. https://doi.org/10.1111/j.1365-2141.2010.08472.x PMid:21275954
- Biernaux
C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl
gene expression at a very low level in blood cells of some healthy
individuals. Blood 1995; 86: 3118-22. https://doi.org/10.1182/blood.V86.8.3118.3118
- Ismail
SI, Naffa RG, Yousef AMF, Ghanim MT. Incidence of bcr-abl fusion
transcripts in healthy individuals. Med Rep. 2014; 9: 1271-6. https://doi.org/10.3892/mmr.2014.1951 PMid:24535287
- Suzuki
T, Kiyoi H, Ozeki K, Tomita A, Yamaji S, Suzuki R et al. Clinical
characteristics and prognostic implications of NPM1 mutations in acute
myeloid leukemia. Blood 2005; 106(8):2854-61. https://doi.org/10.1182/blood-2005-04-1733 PMid:15994285
- Verhaak
RGW, Goudswaard CS, Van Putten W, Bijl MA, Sanders MA, Hugens W et al.
Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML):
Association with other gene abnormalities and previously established
gene expression signatures and their favorable prognostic significance.
Blood 2005; 106: 3747-54. https://doi.org/10.1182/blood-2005-05-2168 PMid:16109776
- Konoplev
S, Yin CC, Kornblau SM, Kantarjian HM, Konopleva M, Andreeff M et al.
Molecular characterization of de novo Philadelphia chromosome-positive
acute myeloid leukemia. Leuk Lymphoma 2013; 54: 138-44. https://doi.org/10.3109/10428194.2012.701739 PMid:22691121 PMCid:PMC3925981
- Reboursiere
E, Chantepie S, Gac AC, Reman O. Rare but authentic
Philadelphia-positive acute myeloblastic leukemia: Two case reports and
a literature review of characteristics, treatment and outcome.
Hematology/ Oncology and Stem Cell Ther. 2014, 8(1):28-33. https://doi.org/10.1016/j.hemonc.2014.09.002 PMid:25300567
- Mariotti
B, Meconi F, Palmieri R, De Bellis E, Lavorgna S, Ottone T et al. Acute
Myeloid Leukemia with Concomitant BCR-ABL and NPM1 Mutations. Case Rep
Hematol. 2019; 6707506. https://doi.org/10.1155/2019/6707506 PMid:31110828 PMCid:PMC6487162
- Castagnetti
F, Gugliotta G, Breccia M, Iurlo A, Levato L, Albano F et al. The
BCR-ABL1 transcript type influences response and outcome in
Philadelphia chromosome-positive chronic myeloid leukemia patients
treated frontline with imatinib. Am J Hematol. 2017; 92(8):797-805. https://doi.org/10.1002/ajh.24774 PMid:28466557
- Neuendorff
NR, Burmeister T, Dörken B, Westermann J. BCR-ABL-positive acute
myeloid leukemia: a new entity? Analysis of clinical and molecular
features. Ann Hematol. 2016; 95: 1211-21. https://doi.org/10.1007/s00277-016-2721-z PMid:27297971
- Jain
P, Kantarjian HM, Ghorab A, Sasaki K, Jabbour EJ, Nogueras Gonzalez G
et al. Prognostic factors and survival outcomes in patients with
chronic myeloid leukemia in blast phase in the tyrosine kinase
inhibitor era: Cohort study of 477 patients. Cancer 2017;
123(22):4391-4402. https://doi.org/10.1002/cncr.30864 PMid:28743165 PMCid:PMC5673547
- Gabert
J, Beillard E, van der Velden VHJ, Bi W, Grimwade D, Pallisgaard N et
al. Standardization and quality control studies of 'real time'
quantitative reverse transcriptase polymerase chain reaction of fusion
gene transcripts for residual disease detection in leukemia - A Europe
Against Cancer Program. Leukemia 2003; 17: 2318-2357. https://doi.org/10.1038/sj.leu.2403135 PMid:14562125
- Ammatuna
E, Noguera NI, Zangrilli D, Curzi P, Panetta P, Bencivenga P et al.
Rapid detection of nucleophosmin (NPM1) mutations in acute myeloid
leukemia by denaturing HPLC. Clin Chem 2005, 51: 2165-2167. https://doi.org/10.1373/clinchem.2005.055707 PMid:16244291
- Gorello
P, Cazzaniga G, Alberti F, Dell' Oro MG, Gottardi E, Specchia G et al.
Quantitative assessment of minimal residual disease in acute myeloid
leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 2006;
20: 1103-1108. https://doi.org/10.1038/sj.leu.2404149 PMid:16541144
- Reid
AG, De Melo VA, Elderfield K, Clark I, Marin D, Apperley J et al.
phenotype of blasts in chronic myeloid leukemia in blastic
phase-Analysis of bone marrow trephine biopsies and correlation with
cytogenetics. Leuk Research 2009, 33: 418-425. https://doi.org/10.1016/j.leukres.2008.07.019 PMid:18760473
- Shah
NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J et al.
Multiple BCR-ABL kinase domain mutations confer polyclonal resistance
to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and
blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2: 117-25. https://doi.org/10.1016/S1535-6108(02)00096-X
- Nacheva
EP, Grace CD, Brazma D, Gancheva K, Howard-Reeves J, Rai L et al. Does
BCR/ABL1 positive Acute Myeloid Leukaemia Exist?. Br J Haematol. 2013;
161: 541-50. https://doi.org/10.1111/bjh.12301 PMid:23521501
- Atfy
M, Al Azizi NMA, Elnaggar AM. Incidence of Philadelphia-chromosome in
acute myelogenous leukemia and biphenotypic acute leukemia patients:
And its role in their outcome. Leuk Research 2011. 35: 1339-44. https://doi.org/10.1016/j.leukres.2011.04.011 PMid:21612824
- Varma
S, Varma N, Malhotra P, Narang V, Sachdeva MU, Bose P.
Immunophenotyping in Chronic Myeloid Leukemia Blast Crisis: Looking
beyond Morphology. J Postgrad Med Edu Res. 2016; 50: 181-184.52. https://doi.org/10.5005/jp-journals-10028-1215
- Soverini
S, Bassan R, Lion T. Treatment and monitoring of Philadelphia
chromosome-positive leukemia patients: Recent advances and remaining
challenges. J Hematol Oncol. 2019;12(1):39. https://doi.org/10.1186/s13045-019-0729-2 PMid:31014376 PMCid:PMC6480772
- Klco
JM, Miller CA, Griffith M, Petti A, Spencer DH, Ketkar-Kulkarni S et
al. Association between mutation clearance after induction therapy and
outcomes in acute myeloid leukemia. JAMA 2015; 314: 811-822. https://doi.org/10.1001/jama.2015.9643 PMid:26305651 PMCid:PMC4621257
- Wang
F, Chan LWC, Tsui NBY, Wong SCC, Siu PM, Yip SP et al. Coexpression
pattern analysis of NPM1-associated genes in chronic myelogenous
leukemia. Biomed Res Int. 2015; 2015: 1-9. https://doi.org/10.1155/2015/610595 PMid:25961029 PMCid:PMC4413041
- Chan
LWC, Lin X, Yung G, Lui T, Chiu YM, Wang F et al. Novel structural
co-expression analysis linking the NPM1-associated ribosomal biogenesis
network to chronic myelogenous leukemia. Sci Rep 2015; 5: 10973. https://doi.org/10.1038/srep10973 PMid:26205693 PMCid:PMC4513283
- Quintás-Cardama
A, Kantarjian H, Cortes J. Basophilic Blast Phase (B-BP) of Chronic
Myelogenous Leukemia (CML). Blood 2007; 110: 4562.
https://doi.org/10.1182/blood.V110.11.4562.4562
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999; 340:1330-40. https://doi.org/10.1056/NEJM199904293401706 PMid:10219069
- Kadam
PR, Nanjangud GJ, Advani SH, Nair C, Banavali S, Gopal R et al.
Chromosomal characteristics of chronic and blastic phase of chronic
myeloid leukemia. A study of 100 patients in India. Cancer Genet
Cytogenet. 1991; 51:167-81. https://doi.org/10.1016/0165-4608(91)90129-I
- Khalidi
HS, Brynes RK, Medeiros LJ, Chang KL, Slovak ML, Snyder DS et al. The
immunophenotype of blast transformation of chronic myelogenous
leukemia: A high frequency of mixed lineage phenotype in 'lymphoid'
blasts and a comparison of morphologic, immunophenotypic, and molecular
findings. Mod Pathol. 1998; 11: 1211-21.
- Branford
S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST et al. Laying the
foundation for genomically-based risk assessment in chronic myeloid
leukemia. Leukemia. 2019; 33: 1835-1850. https://doi.org/10.1038/s41375-019-0512-y PMid:31209280 PMCid:PMC6893870
- Moarii
M, Papaemmanuil E. Classification and risk assessment in AML:
Integrating cytogenetics and molecular profiling. 2017; 2017: 37-44. https://doi.org/10.1182/asheducation-2017.1.37 PMid:29222235 PMCid:PMC6142605
- Patel
JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al.
Prognostic relevance of integrated genetic profiling in acute myeloid
leukemia. N Engl J Med. 2012; 366: 1079-89. https://doi.org/10.1056/NEJMoa1112304 PMid:22417203 PMCid:PMC3545649
- Vitale
C, Lu X, Abderrahman B, Takahashi K, Ravandi F, Jabbour E. t(9;22) as
secondary alteration in core-binding factor de novo acute myeloid
leukemia. Am J Hematol 2015; 90:E211-2. https://doi.org/10.1002/ajh.24143 PMid:26257212 PMCid:PMC4807602
- Salem
A, Loghavi S, Tang G, Huh YO, Jabbour EJ, Kantarjian H et al. Myeloid
neoplasms with concurrent BCR-ABL1 and CBFB rearrangements: A series of
10 cases of a clinically aggressive neoplasm. Am J Hematol. 2017; 92:
520-528. https://doi.org/10.1002/ajh.24710 PMid:28253536 PMCid:PMC5860811
- Lessard
M, Le Prisé PY. Cytogenetic studies in 56 cases with Ph1-positive
hematologic disorders. Cancer Genet Cytogenet. 1982; 5: 37-49. https://doi.org/10.1016/0165-4608(82)90039-5
- Piccaluga
PP, Sabattini E, Bacci F, Agostinelli C, Righi S, Salmi F et al.
Cytoplasmic mutated nucleophosmin (NPM1) in blast crisis of chronic
myeloid leukaemia. Leukemia 2009; 23: 1370-1. https://doi.org/10.1038/leu.2009.95 PMid:19421226
[TOP]