Hemoglobin La Desirade (Hb La Desirade) is an unstable hemoglobin variant characterized by amino acid Alanine (Ala) replacing Valine (Val) at position 129 (H7) in the beta chain. Hb La Desirade exhibits a decreased oxygen affinity and normal heme-heme interaction.[3,4] This variant was found initially in two unrelated black families, the first in association with HbS from Guadeloupe, and the second associated with HbC or beta-thalassemia in a family of Haitian descent living in France.[3] Interestingly, on analysis by standard electrophoresis, it migrates in the same region as normal HbA. This contributes to lacking recognition in some of these cases since, in such cases, the HbA actually represents a combination of HbA and Hb La Desirade together. More recently, this variant was reported as compound heterozygous with other Hemoglobin variants such as Southeast Asian ovalocytosis and Hb Louisville with varying degrees of clinical manifestations.[4,5]
Herein, we describe the clinical and laboratory findings in several Omani Arab families who presented to our service for various reasons, including screening of extended families of the index patients with this hemoglobin variant. This is the first detailed report of patients who were found to have Hb La Desirade trait alone, and, in some subjects, co-associated with the most common Hb variants, namely sickle cell gene and/or alpha thalassemia.[6,7]
Tables 1 and 2 show the demography, clinical features, laboratory and hematological parameters in six subjects who were heterozygous for Hb La Desirade trait and eleven subjects who demonstrated compound heterozygosity for Hb S with Hb La Desirade. They come from five unrelated kindreds of Omani Arab ancestry (Figure 1). The study was approved by the local medical research and ethics committee, and the study was conducted according to the local ethical guidelines after obtaining consent from all five families.
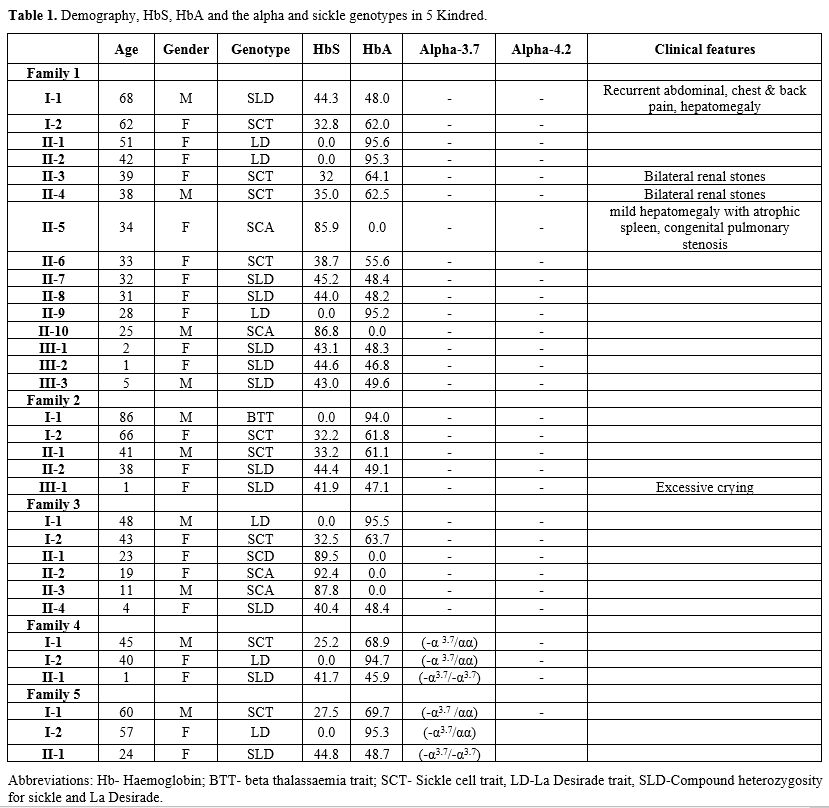 |
Table 1. Demography, HbS, HbA and the alpha and sickle genotypes in 5 Kindred. |
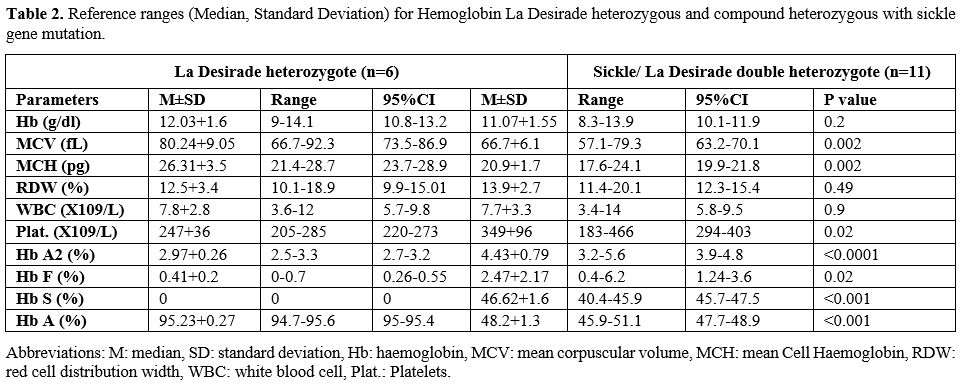 |
Table 2. Reference ranges (Median, Standard Deviation) for Hemoglobin La Desirade heterozygous and compound heterozygous with sickle gene mutation. |
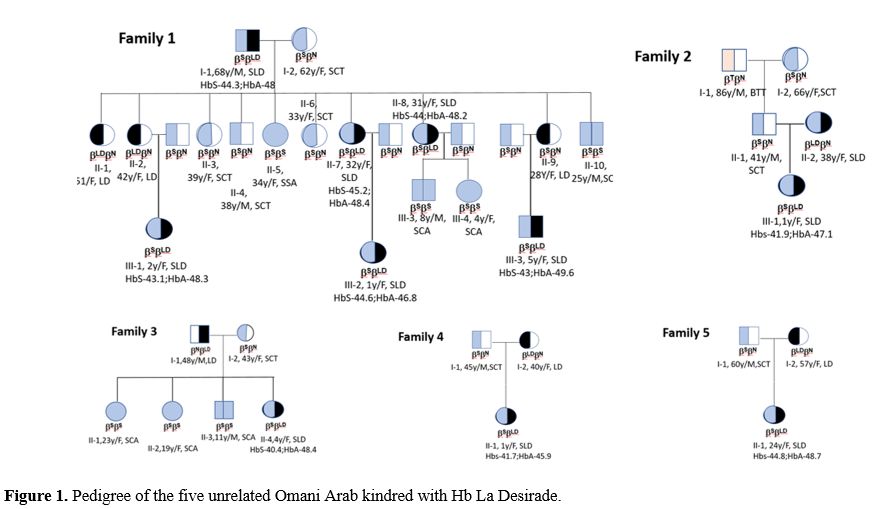 |
Figure 1. Pedigree of the five unrelated Omani Arab kindred with Hb La Desirade. |
Complete blood counts (CBC) were obtained using Cell Dyn 4000 automated blood cell counter (Abbott Diagnostics, Santa Clara, CA, USA) within 4-12 hours of collection, and High-performance Liquid chromatography (HPLC) was done using the Bio-Rad VARIANT ΙΙ™ instrument (Bio-Rad Laboratories, Hercules, CA, USA) using the "β-thalassemia short program". DNA sequencing of the entire β-globin gene segment (including the promoter, all exons, and exon-intron junctions) was performed on an ABI PRISM™ 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Specifically, molecular studies were performed to identify Hb S [β6 (A3) Glu→Val] and Hb La Desirade [β129 (H7) Ala→Val] (Figure 2). Multiplex gap PCR technique was used to detect the common deletional types of alpha thalassemia [Alpha-3.7 and Alpha-4.2].[7]
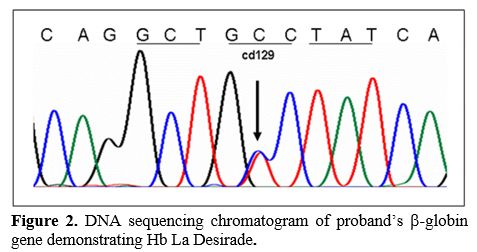 |
Figure 2. DNA sequencing chromatogram of proband’s β-globin gene demonstrating Hb La Desirade. |
In these five kindred, six patients who were found to have Hb La Desirade trait had a mean age of 40.7 years (range 25-57), mean hemoglobin of 12.03 g/dl (range of 9-14.1), mean MCV 80.24 (66.7-92.3) and were clinically asymptomatic. HPLC showed a normal pattern (Figure 3), with a mean HbA of 95.23 % (94.7% -95.6%) and mean HbA2 2.97% (2.5-3.3%). Furthermore, eleven patients with a mean age of 20.5 years (1-68) demonstrated compound heterozygosity for HbS and Hb La Desirade (Figure 3). Their symptoms and signs included recurrent mild painful episodes, including chest and back pain, bilateral renal stones, splenomegaly, and hepatomegaly. A one-year-old child was diagnosed as she presented with excessive crying. Clinically, most of these patients presented and were managed as any other patient with mild sickle cell anemia (SCA). Table 2 indicates the CBC and HPLC findings in patients with Hemoglobin La Desirade heterozygous and compound heterozygous with the sickle gene mutation. The alpha genotype is indicated in Table 1. In addition, within the kindred, there were some normal individuals and several patients with sickle cell trait or sickle cell anemia (Figure 1).
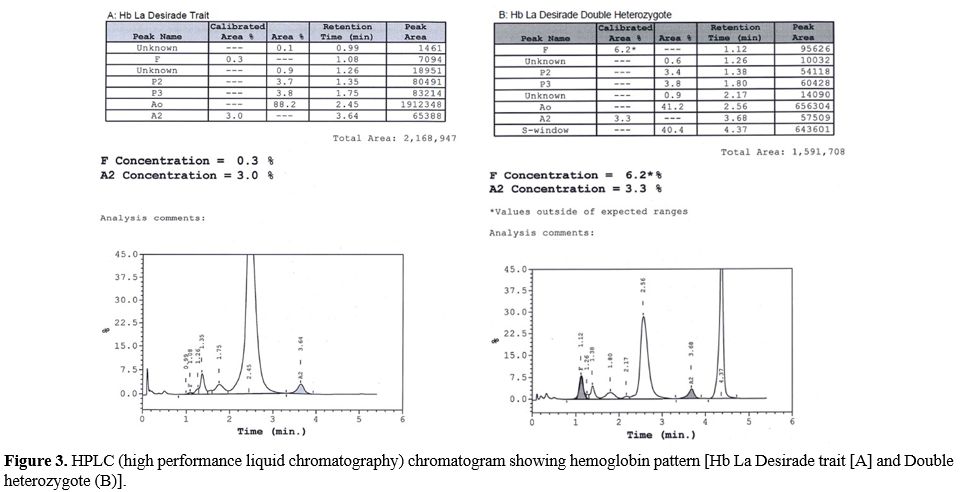 |
Figure 3. HPLC (high
performance liquid chromatography) chromatogram showing hemoglobin
pattern [Hb La Desirade trait [A] and Double heterozygote (B)]. |
Although Hb La Desirade has been reported in population screening, there is little literature on these patients, and this is the first detailed report and insight into the clinical, hematological, and HPLC features of this relatively silent beta hemoglobin variant.[5]
In general, our patients with the Hb La Desirade trait were clinically asymptomatic. Five of the six subjects were females, and their hematological tests were average with no evidence of anemia. Besides, their HPLC also did not show any abnormality with normal HbA, HbA2, and HbF since Hb La Desirade elutes along with HbA (Figure 2). However, its importance is highlighted when associated with other abnormal hemoglobin variants such as HbS, leading to sickle/La Desirade compound heterozygosity, as in our study patients. These patients were seen to present with mild clinical and biochemical manifestations of SCA. Due to widespread screening for hemoglobinopathies at our institution, these patients were identified at an early age, with the median age of the patients in this study cohort being 15.5 years, and half of them were in their first decade. Furthermore, there was mild anemia with a significant hypochromia and microcytosis degree on morphological examination, reflecting the alpha thalassemia gene. The most striking feature was that the levels of HbS and HbA were almost equal on HPLC, and these cases could be misdiagnosed as sickle cell trait (SCT) as sickling is positive in all these patients. However, the levels of Hb S in these compound heterozygotes (40.4-45.9) were higher than normally seen for the diagnosis of SCT in this population. Family screening and molecular studies confirmed the double mutation in codon 6(GAG>GTG) and Codon 129(GCC>GTC) in all these patients [HBB: 20A>T; 389C>T]; it is almost impossible to pick up Hb La Desirade without accompanying family and molecular studies.
A close differential diagnosis that needs to be considered is the β-globin gene promoter (-71 C>T) mutation that has been reported in the Omani population, as it shares the HPLC pattern of the Hb La Desirade trait, with almost equal expression of HbA and HbS.[8] Two significant differences need to be highlighted here that can help to separate the two, although molecular studies would be needed to confirm the correct diagnosis. First, the reported literature shows that Hb S tends to be higher than HbA in HbS/-71(C>T), which is the opposite of what is seen in Hb S/La Desirade subjects (Table 1). Secondly, it has been shown that the -71 C>T mutation is a mild β (+) thalassemic allele, which results in elevated HbA2 levels >3.5%; in the Hb La Desirade trait, the HbA2 is within the normal laboratory range, as seen in our cohort. However, if HbA2 levels are borderline, one should be aware of the factors that could influence HbA2 levels, such as iron status, and HbA2 level should be interpreted in light of RBC indices and iron studies.[9] Therefore, it poses a challenge when doing premarital or community screening, and the definitive answer may require a full family study and appropriate confirmatory molecular studies.
Although this is the largest cohort of Hb La Desirade trait and compound heterozygosity for both Hb La Desirade and HbS reported in the literature, the number of subjects is small, influencing the strength of the statistical analysis.
In conclusion, we report five unrelated kindreds of Omani Arab descent with Hb La Desirade trait and compound heterozygosity for both Hb La Desirade and HbS. Notably, the Hb La Desirade trait is clinically silent and shows no abnormality on HPLC in the heterozygous state. When combined with HbS, it shows a level of HbS of above 40%, prompting further assessment. It is, therefore, HPLC results are not adequate in these situations and warrants caution when counseling patients.