Emine Ikbal Atli1*, Hakan Gurkan1, Engin Atli1, Hakki Onur Kirkizlar2, Sinem Yalcintepe1, Selma Demir1, Ufuk Demirci2, Damla Eker1, Cisem Mail1, Rasime Kalkan3 and Ahmet Muzaffer Demir2. .
1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey.
2 Faculty of Medicine, Department of Hematology, Trakya University, Edirne, Turkey.
3 Faculty of Medicine, Department of Medical Genetics, Near East University, Nicosia, Cyprus.
Correspondence to:
Emine Ikbal Atli, Trakya University Faculty of Medicine, Department of
Medical Genetics, Edirne, Balkan Campus, Highway D100, Turkey. Tel.:
0(284) 235-76-41/2330 Postal code: 22030. E-mail:
emine.ikbal@gmail.com
Published: January 1, 2021
Received: October 1, 2020
Accepted: December 13, 2020
Mediterr J Hematol Infect Dis 2021, 13(1): e2021013 DOI
10.4084/MJHID.2021.013
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Advanced
diagnostic methods give an advantage for the identification of
abnormalities in myeloid malignancies. Various researchers have shown
the potential importance of genetic tests before the disease's onset
and in remission. Large testing panels prevent false-negative results
in myeloid malignancies. However, the critical question is how the
results of conventional cytogenetic and molecular cytogenetic
techniques can be merged with NGS technologies. In this paper, we drew
an algorithm for the evaluation of myeloid malignancies. To evaluate
genetic abnormalities, we performed cytogenetics, molecular
cytogenetics, and NGS testing in myeloid malignancies. In this study,
we analyzed 100 patients admitted to the Medical Genetics Laboratory
with different myeloid malignancies. We highlighted the possible
diagnostic algorithm for cytogenetically normal cases. We applied NGS
141 gene panel for cytogenetically normal patients, and we detected two
or more pathogenic variations in 61 out of 100 patients (61%). NGS's
pathogenic variation detection rate varies in disease groups: they were
present in 85% of A.M.L. and 23% of M.D.S. Here, we identified 24 novel
variations out of total pathogenic variations in myeloid malignancies.
A total of 18 novel variations were identified in A.M.L., and 6 novel
variations were identified in M.D.S. Despite long turnaround times,
conventional techniques are still a golden standard for myeloid
malignancies but sometimes cryptic gene fusions or complex
abnormalities cannot be easily identified by conventional techniques.
In these conditions, advanced technologies like NGS are highly
recommended.
|
Introduction
Myeloid
malignancies originate from hematopoietic progenitor cells and are
characterized by defective differentiation of myeloid progenitor cells.[1]
Advanced molecular detection techniques have changed the diagnostic
algorithm of cancer. Increased next-generation sequencing (NGS) usage
can help change the scope, timing, and suitability of genetic testing
in hematologic malignancies.[2] Despite the advances
in NGS technology and the rising number of study findings that support
the diagnostic and prognostic usage of mutational profiling in
myeloproliferative neoplasms (M.P.N.), the clinical decision-making
role is still not fully utilized.[3]
Diagnostics
algorithms of acute myeloid leukemia (A.M.L.), myelodysplastic
syndromes (M.D.S.), and myeloproliferative neoplasms (M.P.N.) have
evolved in recent years.[4,5] Due to NGS technology
advances, various myeloid NGS panels are commercially available and
generally analyze 25–50 genes classified into several functional
categories including the splicing machinery, epigenetic modifiers, and
transcription factors signaling molecules and chromatin modifiers.[6,7,8,9] The increased knowledge of genetic abnormalities has led to a reclassification of Acute Myeloid Leukaemia (A.M.L.).[10] The World Health Organization[11] and European LeukemiaNet[12]
added new subgroups of diseases, and molecular genetic abnormalities
have also been added in diagnostic criteria. An increased number of
mutational, epigenetic, and expression studies will help identify the
novel markers in myeloid malignancies.
National Comprehensive Cancer Network (NCCN) has added mutations in FLT3, NPM1, CEBPA, and K.I.T. genes to evaluate risk;[13] moreover, the ELN guidelines suggest to add TP53, RUNX1, and ASXL1 mutations in the evaluation of risk. According to some studies, SF3B1, IDH1, and IDH2 should also be included.[3,6,7,14,15]
NGS based myeloid gene panels will help for the identification of
multiple recurrent somatic mutations in many A.M.L. patients, and
additional molecular genetic mutations can be detected in most cases,
even within defined A.M.L. entities.[2] In myelodysplastic syndrome (M.D.S.), NGS allows detecting molecular mutations in approximately 90% of patients.[6,16,17]
As a result, NGS data should be interpreted in the context of other
laboratory findings, including cytomorphology, histopathology,
immune-phenotyping, conventional molecular genetics, cytogenetics, and
clinical diagnostic parameters. In this study, we analyzed 100 patients
referred to Medical Genetics Laboratory with different hematologic
malignancies. We performed conventional cytogenetics, molecular
cytogenetics, and NGS analysis in these cases. According to our
results, we highlighted a possible algorithm for cytogenetically
standard cases.
Materials and Methods
Patient samples.
The present study included 100 patients (52 were male, and 38 were
female) from December 2017 to August 2020. Written informed consent was
obtained from all cases. If patients are under 18 (5 children (≤15
years)), a consent form was signed by a parent and/or legal guardian.
The study was approved by the Ethics Committee of our university and
conducted following the ethical principles established in the
Declaration of Helsinki. The median age of cases was 54 years, ranging
from 1 to 90 years, and there were five children (≤15years) in 100
adults. The distribution of patients was shown in Table 2.
Our cohort consists of 100 patients diagnosed with A.M.L. (61) and
M.D.S. (39). D.N.A. was isolated from bone marrow (QIAamp D.N.A. Blood
Mini Kit (bone marrow = 100) (Qiagen, Germany) and peripheral blood
(MagNA Pure system Roche Diagnostics). D.N.A. was quantified using a
Qubit fluorometer (Thermo Fisher Scientific). The patients who have
normal karyotype and fluorescence in situ hybridization (FISH) report
were enrolled in this study. Patients enrolled in this study were newly
diagnosed. Therefore the treatment protocols were not determined yet.
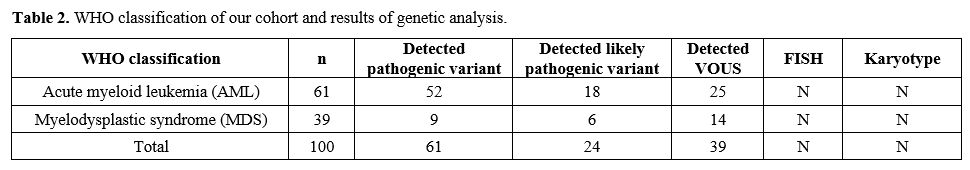 |
Table
2. WHO classification of our cohort and results of genetic analysis. |
Next-generation sequencing.
For evaluating myeloid neoplasm specific 141 genes, the Human Myeloid
Neoplasms QIAseq Targeted D.N.A. Panel (Qiagen, Germany) was used. This
panel covers exon/intron boundaries shown in Figure 1 and covered genes as listed in Table 1.[18]
MiSeq sequencing-by synthesis benchtop sequencer was used for
sequencing of amplified targets according to the manufacturer's
protocol for paired-end sequencing (Illumina, San Diego, CA, U.S.A.).
Data analysis and quality assessment for calling of single-nucleotide
variants and analysis of short insertions and deletions were evaluated
using Ingenuity Variant Analysis (I.V.A.) program. Amplicons were noted
as a dropout and excluded from analysis if the coverage at any analyzed
position in any of the two paired-end sequences (minimal coverage) was
100x, with allele frequency >5% were included for subsequent
investigation. Libraries covering the target genes were prepared
according to the QIAseq Targeted D.N.A. Panel protocol (Qiagen, Hilden,
Germany). Following the target enrichment process, libraries were
sequenced on the MiSeq System and NextSeq 550 System (Illumina, San
Diego, CA, U.S.A.). O.C.I. analysis (Qiagen, Hilden, Germany) was used
for Quality control and Variant Call Format file generation. Variant
analysis has been performed in Ingenuity software (Qiagen, Hilden,
Germany). Variants were interpreted according to the American College
of Medical Genetics, and Genomics 2015 (ACMG-2015) recommended
standards. The candidate variants were annotated by ANNOVAR with SIFT,
PolyPhen-2, MutationTaster, and the Exome Aggregation Consortium (ExAC)
and other databases. Known hotspot or clinically actionable variants
detected below these thresholds were verified using orthogonal methods
such as Sanger sequencing.
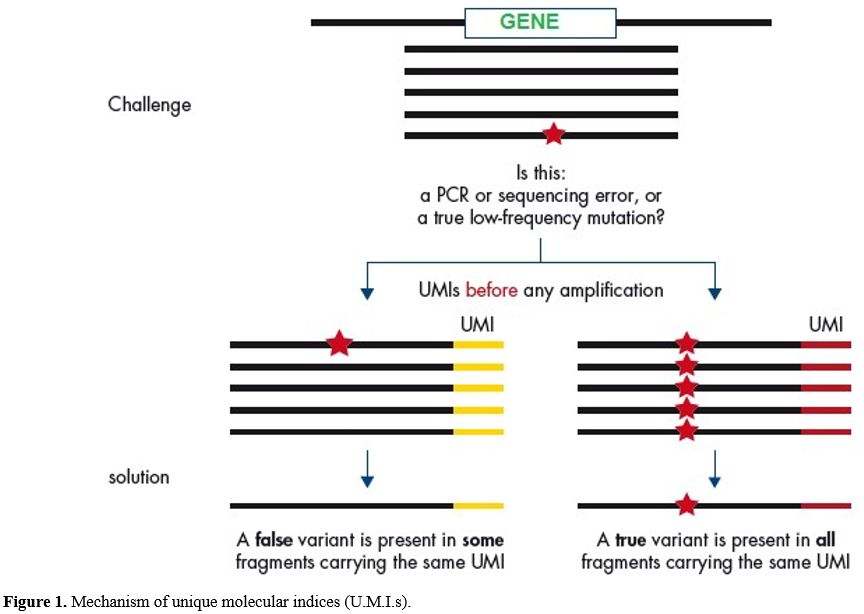 |
Figure
1. Mechanism of unique molecular indices (U.M.I.s). |
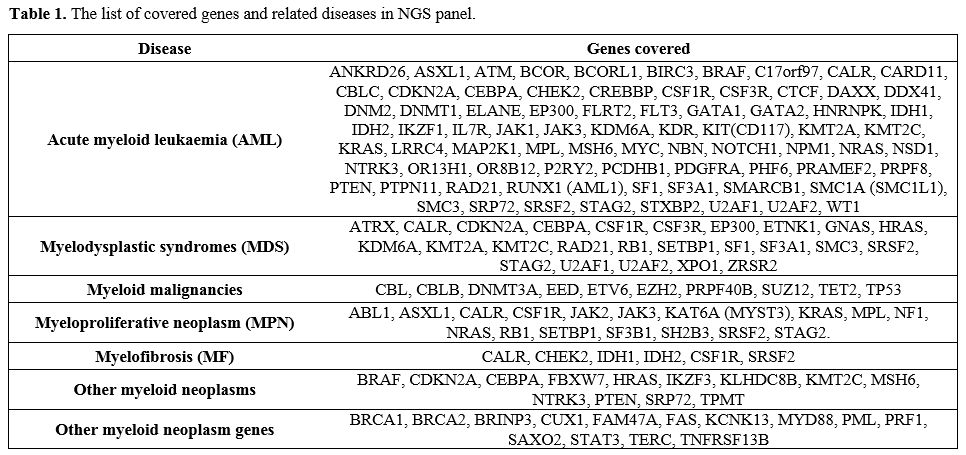 |
Table 1. The list of covered genes and related diseases in NGS panel. |
Cytogenetic Assessment.
Karyotyping: Marrow Max and Chang media were used for cultures of bone marrow and peripheral blood specimens in a CO2
incubator. After 24, 48, or 72 h of incubation, cultures were
harvested. Colcemid was used to arrest metaphase cells, and chromosome
slides were stained using G banding protocol. International System for
Human Cytogenetic Nomenclature (ISCN 2016)[19] was
used for reporting, and 25 metaphases were analyzed in each culture.
The best metaphases were chosen for karyotype analysis, and the total
chromosome count was usually determined in 25 cells.
Fluorescence in situ hybridization (FISH):
FISH was applied according to the manufacturer's recommendations. A
total of 200 interphase cells were analyzed for each sample, and images
were captured/stored by using the Applied Imaging/Cytovision system.
Final results were reported by using the cutoff established in the
laboratory for each of the tested probes.[20] Specific gene panels for FISH was applied for each malignancy. FISH panels for each of the malignancies are listed below.
FISH Panel for AML:
5q-, -5 (5p15, 5q31, 5q33), 7q-, -7 (Cen 7, 7q22, 7q31), Trisomy 8 (Cen
8), MLL (11q23), 20q- (20q12,20qter), RUNX1/RUNX1T1 (ETO/AML1) t(8;21),
PML/RARA t(15;17), CBFB inv(16), t(16;16)
FISH Panel for MDS: 5q-, -5 (5p15, 5q31, 5q33), 7q-, -7(Cen 7, 7q22, 7q31), Trisomy 8 (Cen 8), MLL(11q23), 20q- (20q12, 20qter)
Results
Characteristics of the patients are summarized in Table 2.
Among these patients, 52 were male, and 38 were female. The median age
was 54 years, ranging from 1 to 90 years, and there were 100 adults and
five children (≤15 years).
Results of cytogenetic and molecular cytogenetic analysis. Cytogenetic
and molecular cytogenetic analyses were performed on all of the
patients. Cytogenetics and molecular cytogenetic evaluations were
reported as standard in all of the cases.
Results of next-generation sequencing.
Next-generation sequencing of hotspot regions of 141 genes has been
performed in 100 bone marrow samples referred from the Department of
Hematology. Variables with a depth of coverage > 100x and an allele
frequency of > 5% were included in this study. Known hot spots or
variants identified below the threshold that may require clinical
intervention were confirmed using the Sanger sequencing. Variables of
unknown significance were excluded from the clinical benefit analysis.
Variants were classified as pathogenic and possible pathogenic
according to the gene and clinical effects. Two or more pathogenic
variations were identified in 61 out of 100 patients (61%). A list of
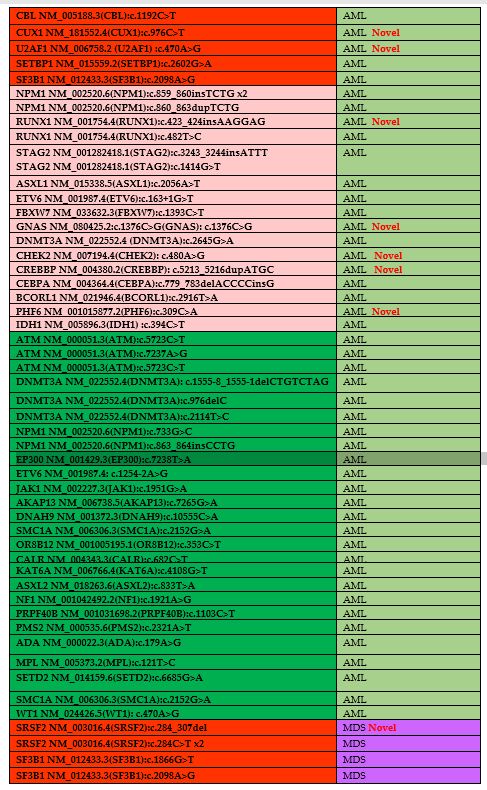
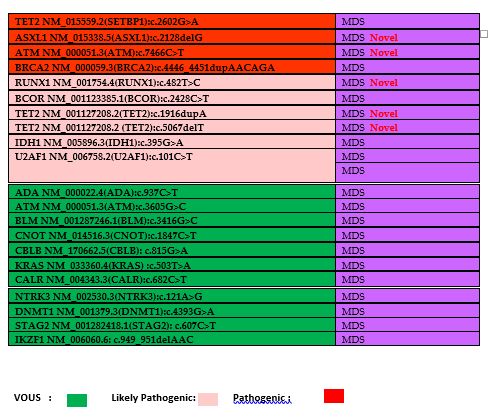
the variants is presented in Table 3.
A total of 24 novel pathogenic or likely pathogenic variations have
been described. In A.M.L., novel pathogenic and likely pathogenic
variations were identified in EP300, STAG2, CUX1, U2AF1, RUNX1, GNAS,
CHEK2, CREBBP, and PHF6 genes. In M.D.S., novel pathogenic and likely
pathogenic variations were identified in SRSF2, ASXL1, A.T.M., RUNX1,
and TET2 genes (Table 3).
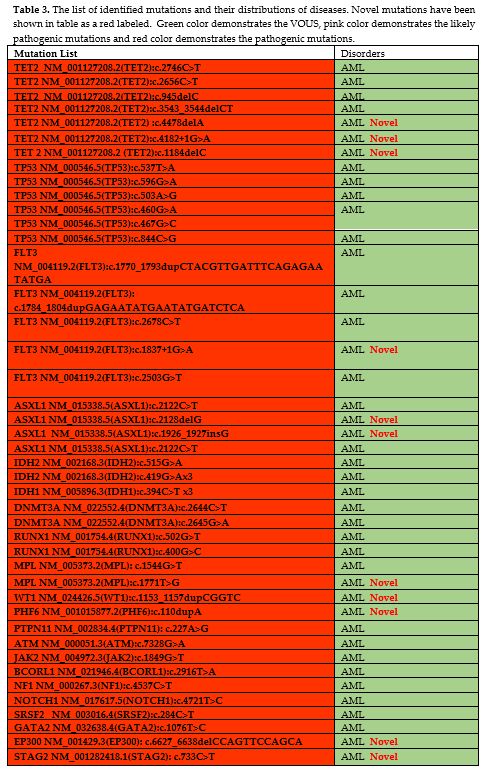 |
Table 3. The list of
identified mutations and their distributions of diseases. Novel
mutations have been shown in table as a red labeled. Green color
demonstrates the VOUS, pink color demonstrates the likely pathogenic
mutations and red color demonstrates the pathogenic mutations. |
 |
|
 |
|
The
distribution of frequent mutations in AML includes, TET2, TP53, FLT3
and IDH2 genes. A total 7 different variants of TET2 (TET2;
c.2746C>T, c.2656C>T, c.945del, c.3543_3544delCT, c.4478delA,
c.1184delC, c.1184delC) were detected in 6 different AML patients and 6
different variants of TP53 (TP53; c.537T>A, c.596G>A,
c.503A>G, c.460G>A, c.467G>C, c.844C>G) were detected in 5
different AML patients. Additionally, pathogenic FLT3 variants
identified in 3 AML patients, including:
c.1770_1793dupCTACGTTGATTTCAGAGAATATGA, c.2503G>T, c.1837+1G>A,
c.2678C>T. The other common pathogenic variants identified in IDH2
gene in AML, including; c.419G>A, c.419G>A and c.419G>A.
The
common pathogenic variants in MDS was SRSF2 and identified in two
different cases, includes: c.284C>T and c.284_307del. More than one
pathogenic variants identified in 2 different cases (Case1: BCOR
c.2428C>T, BRCA2 c.4446_4451dupAACAGA, U2AF1 c.101C>T and case 2:
SRSF2:c.284_307del and IDH1:c.395G>A).
These results show us
that clonality could be observed in the lowest percentages. The
literature recommends that to determine clonally, up to 5% allelic
fraction should be evaluated.[21] NGS's pathogenic
variation detection rate varies in disease groups: in A.M.L. was 85%
(52 out of 61) and M.D.S. was 23% (9 out of 39). Likely pathogenic
variation detection rate and VOUS detection rate of NGS have been
listed in Table 2, and the mutation list of disease groups has been shown in Table 3.
Discussion
Genetic and epigenetic alterations play an important role in leukemogenesis.[22]
Several techniques have been used to identify genetic alterations in
hematologic malignancies, including; FISH, cytogenetics, NGS, RT-PCR
(real time-PCR).[23] Advances in next-generation
sequencing (NGS) technology help transform gene sequencing into a
considerably faster and less expensive test, making it more practical
in clinical practice. The validation of NGS panels is critical, and
generally, a two-step approach is recommended for validation. The first
one is related to the optimization and analysis of relevant errors
during the testing, and the second step is related to the establishment
of thresholds of the depth of coverage and V.A.F. (low variant allele
frequency of variations) for each type of identified variant.[24]
In
recent years, NGS has been used to identify T-cell clonality, recurrent
cytogenetic translocations, and identification of the Philadelphia
chromosome in Acute Lymphoblastic Leukaemia.[2] In
addition to these conditions, in lymphoproliferative diseases, NGS has
also been used to identify clonal I.G.H. and TCR rearrangements in
M.R.D. (Minimal Residual Disease).[21] NGS technology
can be used to identify mutant or clonal D.N.A. in several circulating
tumor cells. It is also essential for clinical trials based
substantially on next-generation sequencing (NGS) parallel it with the
increasing number of molecular markers.[25]
An
increased number of studies in this field will discover new mutations
and update the WHO classification for myeloid malignancies. Moreover,
those studies will help develop novel targeted therapeutic agents and
novel therapeutic targets.[26] Discovering new
mutations in myeloid neoplasms enables us to understand the variable
prognosis and pathogenesis of these diseases. The use of
cytogenetic-based techniques allows identifying "gross" chromosomal
abnormalities such as translocations, amplifications, and deletions.[22]
However, the technique's limitation is based on the abnormality size
because genes can change in various ways (mutations, methylation, etc.)
that may be critical for the onset and/or progression of malignant
hemopathies. The major advancement in NGS is identifying the molecular
basis of leukemia because now we can classify malignant hemopathies at
a molecular level that is more informative than the cytological
classification.[27]
Delic and colleagues
analyzed a 28-gene testing panel in different hematologic malignancies
(myeloproliferative neoplasms, essential thrombocythaemia, primary
myelofibrosis, polycythemia vera). Different mutations were identified
in splicing related genes (SF3B1, SRSF2, and U2AF1), chromatin
modification genes (ASXL1 and EZH2), and methylation related genes
(DNMT3A, IDH1, IDH2, and TET2).[28] Maes et al. analyzed 155 newly diagnosed myeloid neoplasm patients and identified mutation in 81% of the cases.[29]
They highlighted the importance of targeted NGS testing in myeloid
neoplasms' routine diagnostic approach and demonstrates that NGS helps
improve diagnosis, subclassification, and prognosis of cases.[29]
Our
study analyzed 100 myeloid malignancies and identified variations in
61% of cases, and the mutation frequency was similar to the literature.
The critical patient inclusion criterium of the study was the
cytogenetically normal report because we aim to show the importance of
further testing in cytogenetically normal cases during the evaluation
of prognosis of disease and treatment design. Another interesting point
of our study was identifying[24] novel
pathogenic and likely pathogenic variations in myeloid malignancies.
Northrup and colleagues applied a targeted NGS panel to a total of 178
patients diagnosed with myeloid neoplasms. They identified gene
variants in 53% of patients, and they conclude that NGS was a more
sensitive test than conventional cytogenetics, so they suggested that
NGS should become a part of the routine workup of patients.[30] Kawata and colleagues used cytogenetics and NGS for the evaluation of 134 MDS cases.[31]
According to Kawata's work, abnormal NGS was identified in 44 cases
(32.8%). They highlighted together with NGS; the cytogenetic evaluation
also provided more frequent diagnostic information in M.D.S. cases.[31] Studies suggested that NGS can help identify over 80% of recurrent mutations in M.D.S. cases.[8,32]
In our study, NGS's variation detection rate was 61% in myeloid
malignancies, and the detection rate for NGS in M.D.S. was lower (23%)
than what has been described in other studies[8,31,32]
because of a limited number of cases and our inclusion criteria. The
patients who have abnormal cytogenetic reports were excluded from our
study. Because of this reason, our mutation frequency was lower than
the previous studies. Abnormal cytogenetics were closely correlated
with the accumulation of mutations in the transcription factors; cell
cycle checkpoints related genes were associated with normal and
abnormal karyotypes.[33] Therefore, the differences
in variation rates reported in this study were related to our patient
selection criteria, which we enrolled in cases with average karyotype
results. Our present results suggest that NGS could be the right choice
for cases without any cytogenetic alteration, but this approach would
require validation in more extensive studies.
Yu and colleagues
analyzed 43 genes in 93 de novo M.D.S. and 325 non-M3 A.M.L. patients
by NGS and conventional cytogenetics. In 60.1% of cases carries a
complex karyotype, and mutation frequency was detected as 85.8% in
A.M.L. cases.[33] In our study, the detection rate
for NGS in A.M.L. was 85%, which was similar to Yu's study, which
confirms the importance of NGS testing as a diagnostic tool.
Vantyghem
and colleagues conducted a study to show the real-life setting of
chronic myeloid malignancies by NGS testing in a total of 177 chronic
myeloid malignancies patients.[34] They concluded
that NGS's daily practice helps for the final diagnosis of 83% of the
patients.[34] Reinig et al. applied a 42-gene panel in 109 cases of
myelodysplastic syndrome (M.D.S., n: 38), chronic myelomonocytic
leukemia (CMML, n: 14), myeloproliferative neoplasm (M.P.N., n: 24),
and M.D.S. and/or M.P.N. transformed to acute myeloid leukemia (A.M.L.,
n: 33).[35] A pathogenic mutation was identified in 74% of cases of M.D.S., 100% of CMMLs, and 96% of M.P.N.s cases.[35]
Levy and colleagues used a cohort of 380 patients and performed
clinical validation of a gene panel within 50.5% of diagnostic yield.
They concluded that targeted NGS testing should be an alternative to
targeted molecular testing in patients with suspected hematologic
malignancies.[36] Yun et al. used NGS analysis for
evaluation of 157 patients (MDS [n = 95]; secondary-AML (sAML) [n =
52]; CMML [n = 10]) and they highlighted the clinical importance of NGS
during treatment planning of cases.[37] In making the
comparison with our cases, we must make some considerations. We focused
on cases with normal cytogenetic and FISH results, which is a critical
inclusion criterium of patients. All of the cytogenetically abnormal
cases have been excluded from the study. We sequenced 141 genes in a
cohort consisting of 100 patients diagnosed with A.M.L. (61 cases) and
M.D.S. (39 cases). We identified two or more pathogenic variations in
61% of patients. Previous studies aimed to improve NGS usage in all
cases without prior analysis. This study chose the patients who had
normal cytogenetic and FISH results to show the possible false-negative
results depending on the cytogenetic evaluation. Our results also
confirm this hypothesis, showing that those who had normal cytogenetic
evaluation should need further testing by using NGS. We suggested NGS
in routine clinical testing for myeloid malignancies, which are
cytogenetically reported as normal. Here, we identified variations in
different genes related to epigenetic modifications, R.N.A.
modifications, transcription factors, D.N.A. repair, and cohesin
complex. We identified novel variations in EP300, STAG2, CUX1, U2AF1, RUNX1, GNAS, CHEK2, CREBBP, PHF6, SRSF2, ASXL1, A.T.M., RUNX1, and TET2 genes which were not previously described in the literature.
This
procedure will help prevent false-negative results and apply correct
treatment strategies and give prognostic information. Our suggested
algorithm was shown in figure 2, which shows that only cytogenetic analysis is not sufficient to evaluate diseases.
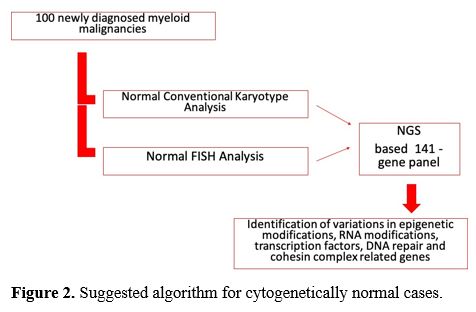 |
Figure 2. Suggested algorithm for cytogenetically normal cases. |
NGS-based
panel testing is widely accepted in clinical practice, and this can
facilitate the construction of well-designed comprehensive NGS panels,
especially during initial diagnosis. Albeit, the targeted NGS panels
can evaluate the genome-wide numerical imbalances. NGS testing gives a
chance to analyze the genomic copy number alteration of interest gene
and which triggers different questions for conventional cytogenetics
during the evaluation of myeloid neoplasms. However, NGS testing cannot
identify structural abnormalities, lacking single-cell resolution, and
low target density, so simultaneous cytogenetic analysis needs to have
a complete picture of the genomic profile. Therefore, after clinical
and diagnostic evaluation, it may be advantageous to perform
cytogenetic analysis for patients whose NGS results show significant
clonal evolution. This procedure has financial consequences, the
requirement of well-trained technical staff, problems during the
bioinformatics analysis of NGS testing. However, despite all of these
conditions, the collected clinical and molecular information should be
led to develop targeted therapeutics in this field.
References
- Visconte V., O Nakashima, M., Rogers, H.; Mutations
in Splicing Factor Genes in Myeloid Malignancies: Significance and
Impact on Clinical Features. Cancers (Basel). 2019 Nov 22;11(12). pii:
E1844. https://doi.org/10.3390/cancers11121844 PMid:31766606 PMCid:PMC6966670
- Palumbo, G.A. et al.; The Role of New Technologies in Myeloproliferative Neoplasms. Front Oncol. 2019 Apr 26;9:321. https://doi.org/10.3389/fonc.2019.00321 PMid:31106152 PMCid:PMC6498877
- Bacher,
U.; et al. Challenges in the introduction of next-generation sequencing
(NGS) for diagnostics of myeloid malignancies into clinical routine
use. Blood Cancer J. 2018 Nov 12;8(11):113. https://doi.org/10.1038/s41408-018-0148-6 PMid:30420667 PMCid:PMC6232163
- Shumilov, E. et al.; Current status and trends in the diagnostics of A.M.L. and M.D.S. Review article. Blood Rev. 2018. https://doi.org/10.1016/j.blre.2018.04.008 PMid:29728319
- Barbui,
T. et al.; The 2016 WHO classification and diagnostic criteria for
myeloproliferative neoplasms: document summary and in-depth discussion.
Blood Cancer J. 8, 2018. https://doi.org/10.1038/s41408-018-0054-y PMid:29426921 PMCid:PMC5807384
- Cazzola,
M., Della Porta, M. G. & Malcovati, L.; The genetic basis of
myelodysplasia and its clinical relevance. Blood 2013,122, 4021-4034. https://doi.org/10.1182/blood-2013-09-381665 PMid:24136165 PMCid:PMC3862275
- Papaemmanuil, E. et al.; Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209-2221.
- Papaemmanuil,
E. et al.; Clinical and biological implications of driver mutations in
myelodysplastic syndromes. Blood 2013,122, 3616-3627.
- Abel,
H.J., Duncavage, E.J.; Detection of structural D.N.A. variation from
next generation sequencing data: a review of informatics approaches,
Cancer Genetics, 206,432- 440, 2013, ISSN 2210-7762, https://doi.org/10.1016/j.cancergen.2013.11.002 PMid:24405614 PMCid:PMC4441822
- Duncavage,
E. J. & Tandon, B.; The utility of next-generation sequencing in
diagnosis and monitoring of acute myeloid leukemia and myelodysplastic
syndromes. Int. J. Lab. Hematol. 37(Suppl 1), 115-121 (2015). https://doi.org/10.1111/ijlh.12361 PMid:25976969
- Arber,
D. A. et al.; The 2016 revision to the World Health Organization
classification of myeloid neoplasms and acute leukemia. Blood 127,
2391-2405 (2016). https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Dohner,
H. et al.; Diagnosis and management of A.M.L. in adults: 2017 ELN
recommendations from an international expert panel. Blood
129:424-447(2017) https://doi.org/10.1182/blood-2016-08-733196 PMid:27895058 PMCid:PMC5291965
- Patel,
U., Luthra, R., Medeiros, L.J., Patel, K.P.; Diagnostic, Prognostic,
and Predictive Utility of Recurrent Somatic Mutations in Myeloid
Neoplasms. Clin Lymphoma Myeloma Leuk. 2017 Jul;17S:S62-S74. https://doi.org/10.1016/j.clml.2017.02.015 PMid:28760304
- Cancer
Genome Atlas Research. Genomic and epigenomic landscapes of adult de
novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059-2074 (2013). https://doi.org/10.1056/NEJMoa1301689 PMid:23634996 PMCid:PMC3767041
- Duncavage,
E. J., Abel, H. J., Szankasi, P., Kelley, T. W., & Pfeifer, J. D.
(2012). Targeted next generation sequencing of clinically significant
gene mutations and translocations in leukemia. Modern pathology : an
official journal of the United States and Canadian Academy of
Pathology, Inc, 25(6), 795-804. https://doi.org/10.1038/modpathol.2012.29 PMid:22425908
- Yoshida, K. et al.; Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64-69(2011)
- Bacher,
U., Kohlmann, A. & Haferlach, T.; Mutational profiling in patients
with M.D.S.: ready for every-day use in the clinic? Best. Pract. Res.
Clin. Haematol. 28,32-42 (2015) https://doi.org/10.1016/j.beha.2014.11.005 PMid:25659728
- A
Sample to Insight®NGS solution for myeloid neoplasms: Redefined
amplicon sequencing for low variant detection and interpretation
(Application Note: PROM-12533- 001)(2018)
- Howe, B., Umrigar, A., Tsien, F.; Chromosome Preparation From Cultured Cells. J. Vis. Exp. (83), e50203.
- Rack,
K.A. et al.; European recommendations and quality assurance for
cytogenomic analysis of haematological neoplasms. Leukemia 33,
1851-1867 (2019) https://doi.org/10.1038/s41375-019-0378-z PMid:30696948 PMCid:PMC6756035
- Gazzola,
A. et al.; The evolution of clonality testing in the diagnosis and
monitoring of hematological malignancies. Therapeutic Adv Hematol.
(2014) 5:35- 47. https://doi.org/10.1177/2040620713519729 PMid:24688753 PMCid:PMC3949299
- De
Braekeleer, E., Douet-Guilbert, N., & De Braekeleer,M.; Genetic
diagnosis in malignant hemopathies: from cytogenetics to
next-generation sequencing, Expert Review of Molecular Diagnostics,
2014, 14:2, 127-129, https://doi.org/10.1586/14737159.2014.872563 PMid:24437978
- Mitelman,
F., Johansson, B., Mertens, F., editors. Mitelman Database of
Chromosome Aberrations and Gene Fusions in Cancer. 2013. Available
from: http://cgap.nci.nih.gov/Chromosomes/Mitelman
- Kim,
H., Yun, J.W., Lee, S.T., Kim, H.J., Kim, S.H., Kim, J.W.; Korean
Society for Genetic Diagnostics Clinical Guidelines Committee. Korean
Society for Genetic Diagnostics Guidelines for Validation of
Next-Generation Sequencing-Based Somatic Variant Detection in
Hematologic Malignancies. Ann Lab Med. 2019 Nov;39(6):515-523. https://doi.org/10.3343/alm.2019.39.6.515 PMid:31240878 PMCid:PMC6660343
- Avila, M., Bernstam, M.; Next-Generation Sequencing for the General Cancer Patient. Clin Adv Hematol Oncol. 2019,17(8):447-454.
- National Comprehensive Cancer Network. Myeloproliferative neoplasms (Version 2.2018). Available from: https://www.nccn.org/professsionals/physician_gls/pdf/mpn.pdf. Accessed September 7, 2017.
- Kuo,
F.C., Steensma, D.P., Dal Cin, P.; Conventional cytogenetics for
myeloid neoplasms in the era of next generation sequencing. AmJHematol.
2017;92:227229. https://doi.org/10.1002/ajh.24642 PMid:28054397
- Delic,
S., Rose, D., Kern, W., Nadarajah, N., Haferlach, C., Haferlach, T.,
Meggendorfer, M.; Application of an NGS-based 28-gene panel in
myeloproliferative neoplasms reveals distinct mutation patterns in
essential thrombocythaemia, primary myelofibrosis and polycythaemia
vera. Br J Haematol. 2016 Nov;175(3):419-426 https://doi.org/10.1111/bjh.14269 PMid:27447873
- Maes,
B., Willemse, J., Broekmans, A., Smets, R., Cruys, B., Put, N., Madoe,
V., Janssen, M., Soepenberg, O., Bries, G., Vrelust, I., Achten, R.,
Van Pelt, K., Buvé, K., Theunissen, K., Peeters, V., & Froyen, G.
Targeted next-generation sequencing using a multigene panel in myeloid
neoplasms: Implementation in clinical diagnostics. Int J Lab Hematol.
2017;39(6):604-612. https://doi.org/10.1111/ijlh.12709 PMid:28722833
- Northrup,
V., Maybank, A., Carson, N., Rahmeh, T.; The Value of Next-Generation
Sequencing in the Screening and Evaluation of Hematologic Neoplasms in
Clinical Practice. Am J Clin Pathol. 2020;153(5):639-645. https://doi.org/10.1093/ajcp/aqz203 PMid:31875888
- Kawata,
E., Lazo‐Langner, A., Xenocostas, A., Hsia, C.C., Howson‐Jan, K.,
Deotare, U., Saini, L., Yang, P., Broadbent, R., Levy, M., Howlett, C.,
Stuart, A., Kerkhof, J., Santos, S., Lin, H., Sadikovic, B. and
Chin‐Yee, I. (2020), Clinical value of next‐generation sequencing
compared to cytogenetics in patients with suspected myelodysplastic
syndrome. Br J Haematol. https://doi.org/10.1111/bjh.16891 PMid:32588428
- Haferlach,
T., Nagata, Y., Grossmann, V., Okuno, Y., Bacher, U., Nagae, G., et
al.; Landscape of genetic lesions in 944 patients with myelodysplastic
syndromes. Leukemia. 2014;28:241-7. https://doi.org/10.1038/leu.2013.336 PMid:24220272 PMCid:PMC3918868
- Yu,
J., Li, Y., Li, T., Li, Y., Xing, H., Sun, H., Sun, L., Wan, D., Liu,
Y., Xie, X., & Jiang, Z. (2020). Gene mutational analysis by NGS
and its clinical significance in patients with myelodysplastic syndrome
and acute myeloid leukemia. Experimental hematology & oncology, 9,
2. https://doi.org/10.1186/s40164-019-0158-5 PMid:31921515 PMCid:PMC6945703
- Vantyghem,
S., Peterlin, P., Thépot, S., Ménard, A., Dubruille, V., Debord, C.,
Guillaume, T., Garnier, A., Le Bourgeois, A., Wuilleme, S., Godon, C.,
Theisen, O., Eveillard, M., Delaunay, J., Maisonneuve, H., Morineau,
N., Villemagne, B., Vigouroux, S., Subiger, F., Lestang, E., … Le Bris,
Y. (2020). Diagnosis and prognosis are comforted by integrated
assessment of next-generation sequencing in chronic myeloid
malignancies. A real-life study. Haematologica, haematol.2019.242677.
Advance online publication. https://doi.org/10.3324/haematol.2019.242677 PMid:32241844
- Reinig,
E., Yang, F., Traer, E., Arora, R., Brown, S., Rattray, R., Braziel,
R., Fan, G., Press, R., & Dunlap, J. (2016). Targeted
Next-Generation Sequencing in Myelodysplastic Syndrome and Chronic
Myelomonocytic Leukemia Aids Diagnosis in Challenging Cases and
Identifies Frequent Spliceosome Mutations in Transformed Acute Myeloid
Leukemia. American journal of clinical pathology, 145(4), 497-506. https://doi.org/10.1093/ajcp/aqw016 PMid:27124934
- Levy,
M.A., Santos, S., Kerkhof, J., et al.; Implementation of an NGS-based
sequencing and gene fusion panel for clinical screening of patients
with suspected hematologic malignancies. Eur J Haematol.
2019;103(3):178-189. doi:10.1111/ejh.13272. https://doi.org/10.1111/ejh.13272 PMid:31177553
- Yun,
S., Geyer, S. M., Komrokji, R. S., Al Ali, N. H., Song, J., Hussaini,
M., Sweet, K. L., Lancet, J. E., List, A. F., Padron, E., &
Sallman, D. A. (2020). Prognostic significance of serial molecular
annotation in myelodysplastic syndromes (M.D.S.) and secondary acute
myeloid leukemia (sAML). Leukemia, https://doi.org/10.1038/s41375-020-0997-4 PMid:32728186
[TOP]