Foluke Atinuke Fasola1, Oluwatoyin Aduke Babalola2, Biobele Jotham Brown3, Abayomi Odetunde2, Adeyinka Gladys Falusi2 and Olufunmilayo Olopade4.
1 Department of Haematology, College of Medicine, University of Ibadan, Nigeria.
2 Institute for Advanced Medical Research and Training, College of Medicine, University of Ibadan, Nigeria.
3 Department of Paediatrics, College of Medicine, University of Ibadan, Nigeria.
4 Department of Medicine, University of Chicago, IL, USA.
Published: January 1, 2022
Received: April 4, 2021
Accepted: November 22, 2021
Mediterr J Hematol Infect Dis 2022, 14(1): e2022001 DOI
10.4084/MJHID.2022.001
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background: Sickle
cell disease is a protean disease with limited data on Nigeria's
phenotypic and genetic variants. This study was conducted to provide
baseline data on these variants by characterising the existing forms of
sickle cell disease and correlating these with basic haematological
parameters.
Methods:
Adult and paediatric patients with SCD were recruited from a tertiary
health centre in Nigeria. Patients were age and sex-matched with
healthy controls. Blood samples were obtained for Full Blood Count,
phenotyping by High-Performance Liquid Chromatography, and genotyping
for alpha thalassemia by multiplex Gap-polymerase chain reaction. Data
analysis was done using IBM SPSS statistics version 23.
Results:
A total of 130 patients with sickle cell disease and 117 controls were
studied. Alpha thalassemia in the study population was due to a 3.7kb
deletion in the alpha-globin gene cluster at a prevalence of 45.4% in
the patients and 47% in the controls. The prevalence of the various
existing forms of SCD genotype was: Homozygous S without alpha gene
deletion (HbSS)- 39.2%; HbSC - 10.8%; HbSSα+1- 35.4%; HbSSα+2 - 6.9% and HbSF- 7.7%. In the control population, HbAA without alpha gene deletion had a prevalence of 42.7%, HbAAα+1 was 25.6%, HbAAα+2 was 6%, HbAS - 7.7%, HbASα+1 – 11.1%, HbASα+2 - 2.6%, HbAC - 2.6% and HbACα+1
- 1.7%. HbA2 was significantly elevated in HbSS individuals with two
alpha gene deletions but reduced in normal controls (HbAA) with alpha
gene deletions. HbF and HbA2 were negatively correlated with each other
(r= -0.587, p < 0.001). Individuals with the HbSC genotype followed
by HbSSα+2 had the best haematological parameters.
Conclusions:
Haematological parameters vary with haemoglobin genotype. The C
haemoglobin and homozygous alpha-thalassemia deletion had a better
ameliorating effect on SCD haematological parameters than the F
haemoglobin in this population. The effect of alpha thalassemia on some
haematological parameters in SCD patients are reversed in normal
controls.
|
Introduction
Sickle
cell disease (SCD) is a heterogeneous group of haemoglobin (Hb)
disorders in which the pathology is attributed to the presence of
sickle haemoglobin (HbS). It is highly prevalent in sub-Saharan Africa.
The heterogeneity in phenotype-genotype has been described, including
homozygosity for HbS represented as HbSS, the double heterozygous
conditions: HbS with HbC disease (HbSC), HbSD Punjab, HbSO-Arab, HbSE
and HbS with different genotypes of beta (β) thalassemia. All these
haemoglobin types are results of mutations in the beta-globin gene.
Other haemoglobins co-inherited with the sickle haemoglobin are fetal
haemoglobin (HbF) and different genotypes of alpha thalassemia.
The
major form of normal haemoglobin in an adult is haemoglobin (HbA), a
tetramer of two alpha (α) globin and two beta (β) globin chains. Normal
minor haemoglobins include HbA2 (alpha and delta chains) and HbF (alpha and gamma chains).
In the normal adult, HbF is usually around 1% and below, while HbA2 is usually about 2%.[1] However, in some SCD patients, the HbF level remains high, and the level of HbA2 may be higher than expected due to the coinheritance of alpha or beta-thalassemia. Haemoglobin F and A2 become diagnostically important when their amount exceeds their reference values in the blood.
The inheritance of alpha thalassemia and HbF with SCD partially explains the clinical and haematological heterogeneity of SCD.[2]
The principal pathophysiology in SCD is HbS polymerisation in the
deoxygenated state resulting in red cell sickling, vaso-occlusion and
haemolysis. The course of disease in patients with SCD is ameliorated
by the inheritance of HbF and alpha thalassemia, earning them the term
genetic modifiers.[3] HbF molecules do not participate
in the polymerisation between molecules of deoxygenated HbS, so
elevated levels of HbF impair the polymerisation of deoxyHbS, resulting
in a milder SCD. Concurrent inheritance of alpha-thalassemia with SCD
reduces intra-erythrocyte HbS concentration, with a consequent
reduction in the polymerisation of deoxyHbS, red cell sickling and
haemolysis. This is particularly important in patients with HbSS, which
is the most common and severe form of SCD.[4]
A range of 0.9 to 16% has been reported for HbF level among our SCD patients using the alkali denaturation technique by Betke.[5,6]
Sickle cell anaemia (HbSS) patients with HbF concentrations greater
than or equal to 20% are compound heterozygotes for HbS and Hereditary
Persistence of Foetal Haemoglobin which can be termed HbS-HPFH, and the
phenotype is represented as HbSF.[7] Patients with HbS-HPFH have mild disease.[8]
A study reported that HbF-increasing β globin gene locus haplotypes
similar to Arab-Indian' and 'Senegal' haplotypes are lacking in
Nigerian patients.[9] The haplotype described in Nigeria is the Benin haplotype.[4] Falusi et al. reported a high prevalence of alpha thalassemia, specifically −α3.7 deletion among Nigerian patients with SCD.[10] These patients with alpha thalassemia also had the tendency to have elevated A2. The degree to which both alpha thalassemia and HbF ameliorate our patients' SCD features is unknown.
SCD patients who inherit a high level of HbF, elevated A2 or alpha thalassemia have distinct haematological features.[11]
The distinct haematological parameter could be an inexpensive tool to
suggest the coinheritance of other haemoglobins, the knowledge of which
could be used to prognosticate patients. Despite the high burden of SCD
in Nigeria, there is limited data on the different forms of the disease
as influenced by the coinheritance of genetic modifiers. The
genotype-phenotype profile of the disease in relation to haematological
parameters is not well defined in many parts of the country. This
limitation is because the diagnosis of SCD using electrophoresis with
cellulose acetate at an alkaline pH as the principal method of routine
screening in our setting has limited the detection of abnormal
haemoglobin phenotypes to HbSS and HbSC and cannot directly quantify
the haemoglobin fractions. The method cannot distinguish HbS from HbD,
HbG and Hb Lepore. Similarly, it cannot distinguish HbC from HbA2, HbE, HbO.[12,13]
HbS and HbC being the most prevalent variant haemoglobins, all
haemoglobins on the location of the band assigned to HbS and HbC are
presumed to be such. The diagnoses of these haemoglobins are usually
validated by the controls (A, S, C) and the presence of sickled
erythrocytes and numerous target cells on peripheral films.
High-performance liquid chromatography (HPLC), the gold standard for
screening and detecting the coinheritance of these haemoglobins, is not
easily accessible in many peripheral health centres, and haemoglobin
genotyping is also not available. HPLC can quantify Hbs F, A2
and S levels to decipher the heterogeneity of SCD further. Hence, this
study aims to use HPLC to determine our SCD patients' haemoglobin
phenotypes and the relationship of the haemoglobin phenotypes and alpha
thalassemia status to the haematological parameters.
Materials and Methods
This is a cross-sectional study of patients diagnosed with SCD by haemoglobin electrophoresis at alkaline pH 8.6[14]
and managed for SCD at the Pediatric Outpatient Clinic and Hematology
Clinic of the University College Hospital, Ibadan. Age- and sex-matched
controls, including school children, students of tertiary institutions
and other individuals from the community, were recruited. The sample
size was 130 for patients and 117 for controls. All patients with SCD
attending the clinic in a steady state and who had not received a blood
transfusion in the last four months preceding the study were eligible
and invited to participate. This exclusion was to avoid the analysis of
haemoglobin fractions from transfused blood in the participants. The
SCD patients were recruited into the study irrespective of whether they
were on hydroxyurea therapy or not. Informed consent was obtained from
both patients and controls: for children, informed consent was obtained
from their parents, including assent from children aged 13-18 years.
Questionnaires were administered to obtain information on demographic
data. Venous blood was collected from each patient and control. Three
milliliters was put in Ethylenediaminetetraacetic acid (EDTA) bottle
for full blood count, high-performance liquid chromatography (HPLC)
analysis and DNA extraction.
Samples were transported in a cold
box from the collection site to the Genetics and Bioethics Research
Unit laboratory, IAMRAT, College of Medicine, University of Ibadan,
Nigeria. High-performance liquid chromatography (HPLC) by the Bio-Rad
Variant IITM HbA2/HbA1C
Dual Program (Clinical Diagnostics, California, USA) was used as
described in the instruction manual for the assay to identify and
quantify HbA, HbS, HbC, HbF and HbA2 in both SCD patients and controls.[15]
The HPLC consists of 2 methods, of which the Beta-thal (HbA2) method
was used for analysis. Full blood count for SCD and controls was
determined using the Swelab 3-part Haematology Analyser (Boule
Diagnostics, Sweden). DNA extraction from whole blood was carried out
at the Genetics and Bioethics Research Unit laboratory, IAMRAT, College
of Medicine, University of Ibadan, Nigeria using Qiagen kit (QIAGEN
Inc., MD, USA) according to manufacturer's instruction.[16]
DNA analysis to determine deletion the alpha-globin gene cluster was carried out
at the Haematology-Oncology Laboratory, Department of Medicine,
University of Chicago. The deletion in the alpha globin gene cluster
was determined by Multiplex gap-PCR.[17] The
participants were identified as heterozygotes when there was only one
gene deletion (−α3.7/αα) represented as α+1, homozygotes are when there
were two gene deletions (−α3.7/−α3.7), represented as α+2. Ethical
approval for this study was obtained from the University of
Ibadan/University College Hospital, Ibadan Institutional Ethics Review
Committee, College of Medicine, University of Ibadan and the
Institutional Review Board at the University of Chicago
Statistical Analysis.
Data were processed using the IBM SPSS statistics version 23. Variables
were tested for normality using the Shapiro-Wilk test. Descriptive
statistics of continuous variables were obtained using mean ± SD,
median and interquartile range (IQR) and categorical variables reported
in proportions. Student T-test was used to compare means between
groups, and Mann-Whitney U test was used to compare median between
groups. Age and hydroxyurea use were controlled for using ANCOVA in the
comparison of haematological parameters in SCD patients. Pearson's
correlation was used to test for a relationship between continuous
variables. Statistical significance was set at p<0.05 (2-tailed).
Sickle cell anaemia patients without alpha thalassemia and HbF lower
than 20% were identified as HbSS, while patients who were not on
hydroxyurea and had HbF ≥20% were categorised as having HPFH, thereby
labelled as HbSF. HbA2 values of 3.5% and above was considered elevated.[12]
Results
Demography of Study Population.
A total of 130 children and adults diagnosed with sickle cell disease
(SCD) and 117 age and sex-matched controls without sickle cell disease
were enrolled in the study. The median (interquartile range) age among
SCD cases was 16 (9 - 29) years with a minimum age of 3 years and a
maximum age of 63 years. Characteristics of the study participants are
shown in Table 1. Twenty (15.4%) of the SCD patients in this study were on hydroxyurea.
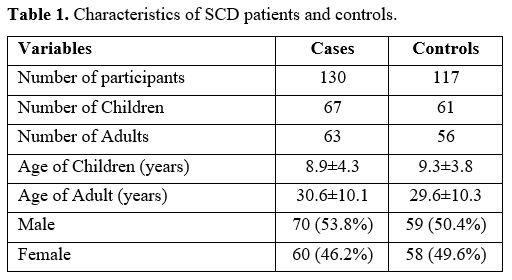 |
Table 1. Characteristics of SCD patients and controls. |
Distribution of Haemoglobin Phenotypes in the Study Population.
Based on the results obtained from HPLC, 106/130 (81.5%) of the SCD
patients were HbSS, 14/130 (10.8%) were HbSC, and 10/130 (7.7%) were
HbSF. The HbSF comprised 7(10.4%) out of 67 children (<18 years) and
3 (4.8%) out of 63 adults (> or = 18 years). All patients who were not on
hydroxyurea with HbF level > 20% were categorised as HbSF. For the
control group, 87/117 (74.4%) were HbAA, 25 (21.4%) were AS, 5(4.3%)
were HbAC.
Comparison of Haematological Parameters Between SCD Population and Controls.
Participants without SCD had statistically significant higher HGB
(Median = 12.60 g/dL), RBC (Median = 4.62x1012), HCT (Median = 40.20%), and LYMF
(Mean = 54.01±11.42%) when compared with SCD patients: HGB (Median =
8.0 g/dL), RBC (Median = 2.79x1012), HCT (Median = 25.0%), and LYMF (Mean =
46.07±12.17%) respectively. The RDW (Median = 22.55%), MCH (Mean =
28.17±3.48 pg), WBC (Median = 10.55x109) GRAN (Mean = 46.85±13.0%) and PCT
(Median = 0.28%) were statistically significantly higher in SCD patients
than among the control group: RDW (Median = 15.95%), MCH (Mean =
27.26±2.94 pg), WBC (Median = 5.30x109), GRAN (Mean = 39.25±13.70%) and PCT
(Median = 0.20%) respectively (Table 2).
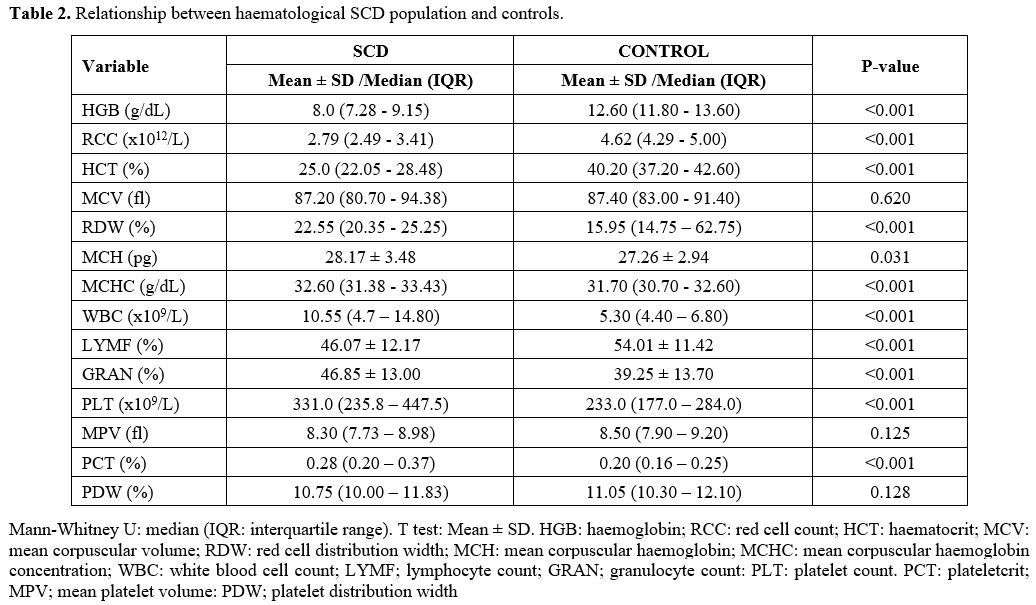 |
Table
2. Relationship between haematological SCD population and controls. |
Alpha Thalassemia in the Study Population.
Alpha thalassemia in the study population was due to a 3.7kb α-globin
gene deletion. Alpha thalassemia was present at a prevalence of 45.4%
(59/130) in the patients and 47% (55/117) in the control group. Based
on the combination of results from HPLC and analysis to detect alpha
thalassemia, the SCD patients were divided into five groups. These
included homozygous S without alpha gene deletion (HbSS), haemoglobin S
with haemoglobin C disease (HbSC), haemoglobin S with coinheritance of
alpha thalassemia with one gene deletion (heterozygous deletion)
otherwise known as silent alpha thalassemia (HbSSα+1), haemoglobin S
with coinheritance of alpha thalassemia with two gene deletions
(homozygous deletion) otherwise known as alpha thalassemia trait (HbSSα+2)
and haemoglobin S with hereditary persistence of foetal haemoglobin
(HbSF). The prevalence of the various existing forms of SCD genotype
was: Homozygous S without alpha gene deletion (HbSS)- 39.2%; HbSC -
10.8%; HbSSα+1- 35.4%; HbSSα+2
– 6.9% and HbSF- 7.7%. Of the 59 patients with alpha thalassemia, 6
(10.2%) had HbA2 ≥3.5%. Four of the SCD patients with α+1 deletion also
had HbC and were categorised as HbSC for analysis. Table 3
shows the distribution of sickle cell patients in each category. In the
control population, HbAA without alpha gene deletion had a prevalence
of 42.7%, HbAAα+1 was 25.6%, HbAAα+2 was 6%, HbAS- 7.7%, HbASα+1 –
11.1%, HbASα+2 - 2.6%, HbAC – 2.6% and HbACα+1 – 1.7%.
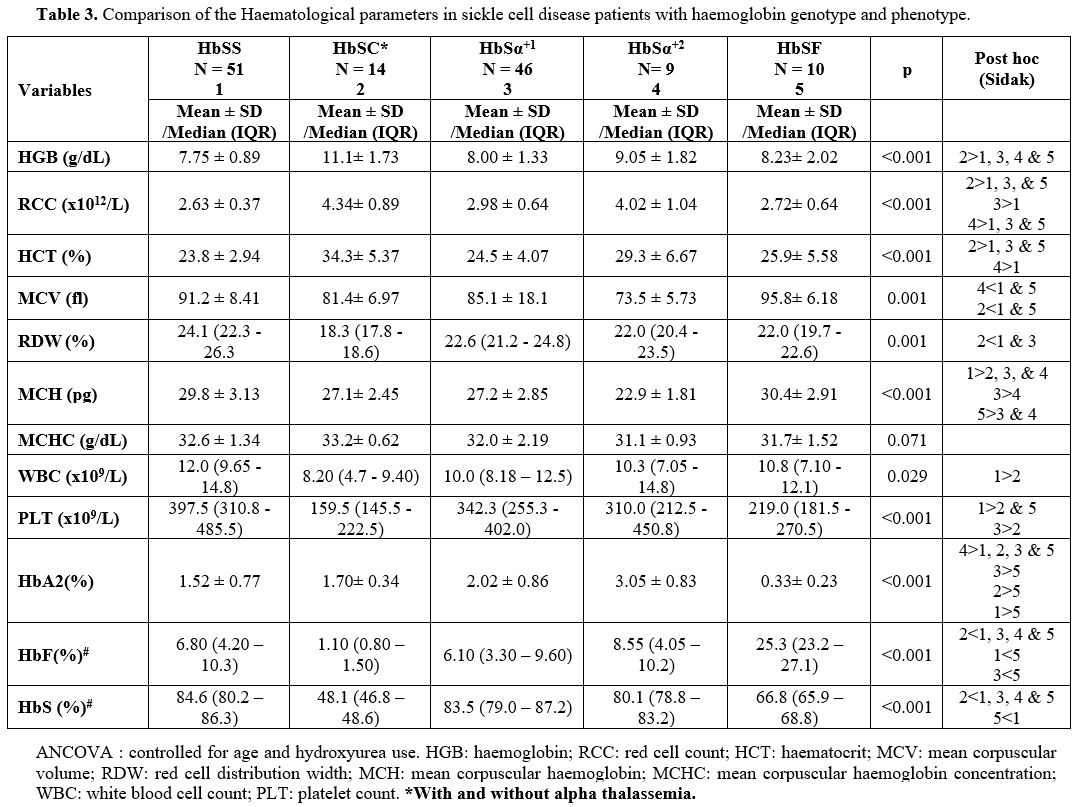 |
Table 3. Comparison of the Haematological parameters in sickle cell disease patients with haemoglobin genotype and phenotype. |
Haematological Parameters for the Sickle Cell Genotype Categories. Table 3
shows the haematological parameters for the different genotype
categories, controlled for age and hydroxyurea use. There was marked
variability of the haematological parameters among the various sickle
cell disease genotypes. Patients with HbSC had the highest haemoglobin
(HGB), haematocrit (HCT) and red cell count (RCC) and the lowest red
cell distribution width (RDW). RDW was similar for HbSα+1, HbSα+2
and HbSF. Homozygous S patients without alpha thalassemia and with HbF
lower than 20% (HbSS) had the lowest HB, HCT, RCC and highest white
blood cell count (WBC), platelet count (PLT) and RDW. The differences
in WBC and PLT were significant only between SS and SC. The white cell
count in HbSSα+2, HbSSα+1
and HbSF were comparable. Platelet count was also significantly higher
in SS than in SF. Mean Corpuscular Volume (MCV) and Mean Corpuscular
Haemoglobin (MCH) were lowest for HbSSα+2 followed by HbSSα+1,
and SC and highest for SS and SF (p<0.001). There was no significant
difference in the MCV between HbSS without alpha thalassemia and HbSF
individuals. There was no significant difference in the Mean
Corpuscular Haemoglobin Concentration (MCHC) among the different SCD
genotypes. Sickle cell disease patients who were on hydroxyurea had
significantly higher MCV (Median = 92.30, IQR = 88.05; 99.03) than SCD
patients who were not on hydroxyurea (Median = 86.90, IQR = 79.83;
93.28) p=0.014. Similarly, there was a statistically significant
difference in MCH between SCD patients on hydroxyurea (29.9 ± 3.26) and
SCD patients not on hydroxyurea (28.0 ± 3.44) p= 0.040.
The mean HbS percentage was significantly lower in SF than in the HbSS, HbSSα+1 and HbSSα+2 (Table 3). The HbA2 level was lowest (0.33 ± 0.23%) in HbSF and highest (3.05 ± 0.83%) in HbSSα+2 (p<0.001). Among the groups of patients, HbF level was lowest in HbSC and comparably low in HbSS, HbSSα+1 and HbSSα+2 (Table 3).
Haematological Parameters for the Control Group.
In the control population, the HGB and HCT concentration reduced with
decreasing number of alpha chains, while RCC increased. These, however,
did not reach statistical significance (Table 4). The difference in the MCV among HbAA controls (89.8 ± 6.27 fl), HbAS controls (84.8 ± 6.92 fl), HbAAα+1 patients (83.6 ± 11.3 fl), HbAAα+2
patients (75.8 ± 4.29 fl), and HbAC controls (81.3 ± 5.08 fl), were
statistically significant, p <0.001. For adequacy of statistical
analysis, the HbAS and HbAC controls could not be separated into those
with and without alpha thalassemia. Thirteen HbAS (11.1%) individuals
had single deletion, and three (2.6%) had double deletion. Two of the
five HbAC individuals had single alpha deletion. MCV in both HbAAα+1 and HbAAα+2
controls were statistically significantly lower than in HbAA (p=0.008
and p<0.001), respectively. However, the mean MCV for AS and AC did
not differ significantly from other groups, respectively. MCH level in
HbAAα+2 controls was significantly lower than in AAα+1 (p=0.049), HbAS (p=0.006) and AA (p<0.001) respectively. The result also showed that the MCH level in HbAAα+1 controls was significantly lower than in AA controls (p=0.001).
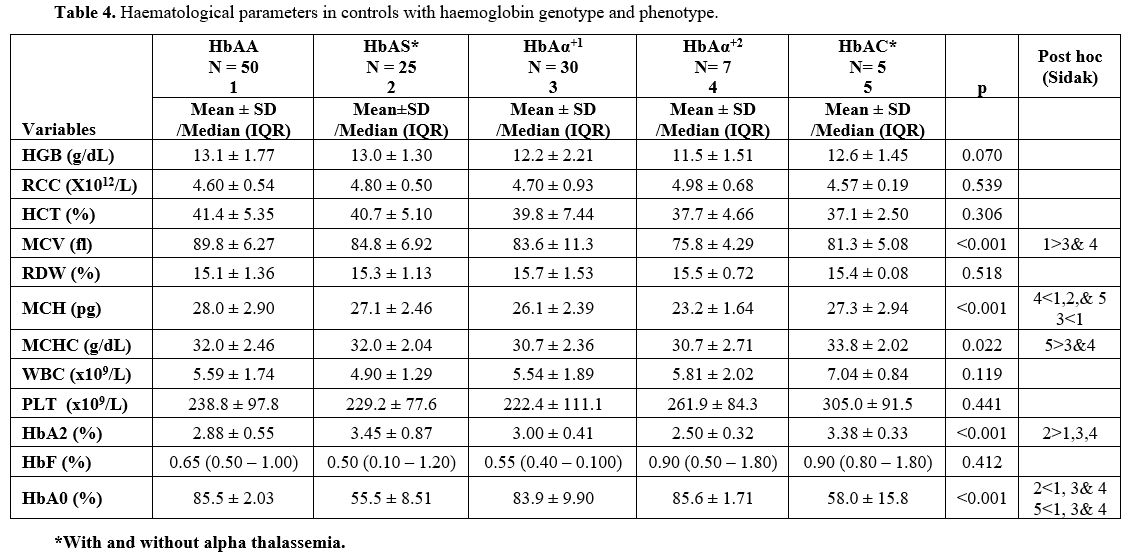 |
Table 4. Haematological parameters in controls with haemoglobin genotype and phenotype. |
Haemoglobins F and A2 in the Study Population.
The mean HbF level in the patients that were not on hydroxyurea was 8.3
± 7.3% (0.2 - 30.6%), while the mean HbF level of patients on
hydroxyurea was 9.9±7.3% (1.8-25.1%). There was a negative correlation
between HbF level and age in patients not on hydroxyurea (r= -0.260, p=
0.009). All the different categories of SCD have varying levels of HbF
irrespective of the alpha thalassemia status. Excluding patients on
hydroxyurea, 29/110 (26%) patients had HbF level >10%. The mean HbA2
level in the SCD patients was 1.71 ± 0.95% with a range of 0.1 - 4.0%,
while the mean HbA2 level in controls was 3.03 ± 0.63% with a range of
0.9 - 4.42%. The level of HbA2 (%) in HbAS controls was significantly
higher than in HbAA (p=0.002), HbAAα+1 (p=0.029) and HbAAα+2 (p=0.005) respectively (Table 4).
Six (10.7%) of the SCD patients with alpha thalassemia had HbA2 ≥3.5%,
while 22 (40.0%) controls with alpha thalassemia had HbA2 ≥3.5%. Very low HbA2 were observed in some patients with HbSF, HbSC, HbSS without alpha thalassemia.
Discussion
This
study shows the heterogeneity of sickle cell disease among our patients
and attempts to relate it to haematological indices. Homozygous S
(HbSS) patients constituted 81.5% of our patients, which is lower than
90% reported among SCD in the Northern part of the country.[18] and then 92.1% of a previous study among children in our centre.[19]
The disparity in the prevalence of SS between this study and the
previous study in our centre might be attributed to the separation of
HbSF from HbSS in the current study, unlike in the previous study. The
dissimilarity with this study conducted in South Western Nigeria from
the study in Northern Nigeria may reflect the lower prevalence of HbSC
(4%) in Northern Nigeria[18] compared to South
Western Nigeria with higher HbSC mutation (10.8%) as reflected in our
study. This lower prevalence in the Northern part of the country has
also been documented by Akinyanju et al.[20] The prevalence of SC in our study is consistent with Brown et al.[21].
The documentation on the prevalence of HbSF in Nigerian patients with
SCD is scanty. A prevalence of 4.8% among our adult patients is higher
than 2% reported for adult patients in Lagos by Akinbami et al.,[22]
but this disparity may not be significant considering the sample size
of the adult patients in both studies. A prevalence of 3.2% in 180
pediatric patients in Lagos reported by Adeyemo et al.[23]
is much lower than the 10.4% of the 67 paediatric patients of this
study. Factors influencing the behavior and decision of patients to go
to the hospital can contribute to variation in prevalence rate in
hospital-based studies. In line with those mentioned above, most
studies on the prevalence of SF have been hospital-based, leaving out
SCD patients with a mild clinical course and less likely to be followed
up in the Haematology clinic. If the patients were eventually
registered in the clinic, they might visit the hospital infrequently
depending on how conducive and convenient the hospital environment is.
This behavior may cause variability in reports from hospital-based
studies. Since the health system in Nigeria is pay out of pocket, SCD
phenotypes with less severe disease manifestations are also less likely
to be encountered in a hospital-based study, except they are
financially buoyant. HbSF was not detected in a survey of 10,001
infants and children from the Northern part of the country.[19]
This finding underscores the variation in the geographical distribution
of Hb phenotype within the same country and the high variability of HbF
level in the Benin haplotype seen in the West African population where
Nigeria belongs.[22] The possibility of the spread of
the Bantu haplotype with the least HbF level to Northern Nigeria being
close to the Cameroon region should be considered since the haplotype
in the study participants was not investigated.
Alpha
thalassemia reduces the concentration of HbS in the red cell of the
patients; however, the consequence of the reduction varies in different
populations. In the Democratic Republic of Congo, among pregnant women,
morbidity related to sickle cell complications in the mother and foetus
were less frequent in the HbSS/alpha-thal than in HbSS.[24] The protective role of α-thalassemia against the risk of abnormal TCD in Nigerian children with SCA has been described.[25] The coinheritance of alpha thalassemia and SCA has also been associated with lower consultation rates and increased survival.[26] However, a frequent and severe vaso-occlusive crisis has been associated with alpha thalassemia in children.[27]
A significant proportion (45.4%) of our SCD patients had
alpha-thalassemia due to 3.7kb α-globin gene deletion, suggesting
heterogeneity in the sickle cell genotype and supporting the report of
the previous study carried out by Falusi et al. in the same centre[10] and in blacks.[28,29]
Of all the SCD patients with alpha thalassemia, the ameliorating effect
may be expected in the 15.3% with homozygous gene deletion because the
single gene deletion has minimal effect on the pathophysiology of
sickle cell disease.[30] The haematological
parameters in our patients varied with the haemoglobin phenotypes and
genotypes, further underscoring the significance of haematological
parameters in categorising the patients. Patients with HbSC had the
highest HGB level, RCC and HCT, followed by patients with homozygous
alpha-thalassemia and then HbSF. Red cell count was significantly
higher in homozygous alpha-thalassemia than in heterozygous
alpha-thalassemia; the higher HGB and HCT was not significant. Higher
HGB and RCC in patients with alpha thalassemia is due to a reduction in
the rate of hemolysis[31] and increased red cell precursor mitosis, smaller red cells (low MCV) and relative erythrocytosis for the degree of anaemia.[32]
Our finding corroborates the previous report about high haemoglobin in
patients with α-thalassemia, which is related to the number of alpha
deletions.[33]
White blood cell count (WBC) and
platelet count (PCT) have been documented to be higher in patients with
SCD compared with their healthy counterparts.[34] This is consistent with the findings in our study with mean WBC and platelet count of 10.5x109/L and 331.0 x 109/L respectively in SCD compared to 5.30x109/L and 233x109/L
in the non-SCD controls. There are multiple causes for leukocytosis in
SCD. The most important reason is the marked inflammation that
characterises the disease. In addition to inflammation contributing to
a higher platelet count in SCD than in controls, auto splenectomy could
also contribute to the elevated platelet count. In agreement with
reports from other studies,[31,35]
the mean HGB, HCT, and RCC counts of the patients showed gradual
increment from no alpha deletion, through one alpha deletion to 2 alpha
deletions; while MCV, MCH, RDW, MCHC, WBC, PLT and HbS level showed a
gradual decrement in the same direction. This shows a reversal of the
expected high WBC and PLT in patients with SCD. Also, significant
gradual decrement is observed in controls' MCH and MCV values with the
number of alpha deletions. Haemoglobin concentration and haematocrit
increased with the number of alpha genes deleted in the patients,
unlike in the control group, where the reverse was the case. The degree
of anaemia, leukocytosis and platelet count due to alpha thalassemia in
the patients may be reduced because the severity of the disease is
ameliorated by reducing the amount of sickled RBC, the intracellular
HbS level and inflammatory process. All these lower cell damage and
improve the hemolysis profile. The inheritance of 2 alpha deletions
together with HbSS in our study population had a more identifiable
haematologic phenotype than the inheritance of one alpha deletion with
HbSS. In this population of patients, homozygous alpha thalassemia has
a better ameliorating effect on haematological parameters than fetal
haemoglobin. An explanation for the lower levels of haemoglobin,
hematocrit and red cell count in HbSF is that HbF level per F cell
(HbF/F-cell) is the critical predictor of the impact of HbF on the
manifestation of disease and not the absolute level of HbF in the
blood.[36] The effect of the Benin haplotype on HbF
distribution within the cells is not known. The distribution of HbF
among F cells determines its ability to inhibit HbS polymerisation.
Patients with high HbF can have a severe disease if HbF is unevenly
distributed among F-cells. The uneven distribution of HbF may result in
some cells having insufficient concentrations to inhibit HbS
polymerisation.[36,37] This suggests that certain
patients may not benefit from the use of HbF inducing agents such as
hydroxyurea despite attaining a "threshold" HbF value of >20%, which
is the protection level for sickle cell-related morbidity.[37]
The
red cell distribution width (RDW) was high for all Hb phenotypes. It
was highest in HbSS without alpha deletion and least in HbSC. High RDW
is associated with impaired erythropoiesis and abnormal red blood cell
survival.[38] This RDW may reflect the order of
severity of the disease as an increased RDW has a high negative
predictive value for diagnosing a variety of disorders and also gives
information as to prognosis.[39] Low red cell count
with high RDW and high normal platelets may suggest an iron deficiency
in the HbSS without alpha deletion, but the normal MCV, MCH and MCHC
make the diagnosis unlikely even though iron studies were not carried
out on the participants. Mean corpuscular haemoglobin concentration
(MCHC) is assumed to be a determinant of disease severity;[40]
however, it might not play this role among our patients as it does not
show significant difference among the various Hb phenotypes of our
patients. Despite the non-significant difference among the various Hb
phenotypes, HbSα+2 had the lowest MCHC. However, our finding was consistent with a report by Velasco-Rodríguez et al.,[40]
that MCH and MCV stood out as the most efficient red cell indices to
predict the deletion of two alpha genes. Also, our findings
corroborated with documentation by Old[41] that the
MCHC is normal in heterozygous coinheritance with thalassemia while the
MCV and MCH are significantly lower than in SCD without alpha
thalassemia. Since there was no significant difference in the MCHC for
all the Hb phenotypes in this group of patients, MCHC may not be useful
to differentiate the various Hb phenotypes; rather, MCV and MCH could
be used.
Interestingly, in the control population, MCHC is
significantly higher in HbAC individuals than those with homozygous and
heterozygous alpha thalassemia. The explanation lies in the fact that
the rigidity and dehydration of this abnormal haemoglobin cause
microcytosis and compacted normal amounts of haemoglobin. Though MCV
and MCH were lower in SCD with homozygous alpha-thalassemia than in
heterozygous alpha thalassemia, only MCH showed a significant
difference between the two disorders, suggesting a low MCH is a
stronger indicator of the severity of the thalassemia than low MCV.
Mean corpuscular haemoglobin concentration is traditionally calculated
by dividing the haemoglobin quantity by the hematocrit. The advent of
automated haematology analysers for routine haematology investigation
has caused a shift of emphasis from MCHC, which is affected by the
methodology used to derive value.[42] Therefore, MCHC is mostly normal in microcytic anaemias while the value of MCH closely parallels the value of MCV.[43]
This
study shows that HbS patients with alpha thalassemia have varying
levels of HbF. Iron deficiency anaemia and inheritance of thalassemia
could affect the correlation between HbF levels and red cell indices.[43]
Higher MCH and MCV levels in HbSS without alpha thalassemia and HbSF
individuals than SCD individuals with heterozygous and homozygous
alpha-thalassemia are expected. This is due to the lower amount of
alpha chains in the red blood corpuscles of patients with alpha
thalassemia. The higher MCH and MCV in non-thalassemic individuals
relative to alpha thalassemia presence is also observed in normal
controls. The contribution of folate deficiency to the MCV value could
not be ruled out in these patients whose compliance to the prescribed
routine folic acid was not investigated, but the MCV of less than 100fl
in the patients makes it unlikely.
The observation of a higher HbA2 level in patients with alpha thalassemia than in those without corroborates the findings of previous studies by other authors.[11,27] However, considering the range of the HbA2 values in our thalassemic patients, it can also be inferred that the absence of elevated A2 does not necessarily rule out alpha-thalassemia in patients with SCD, even if elevated HbA2 may be suggestive of alpha thalassemia in SCD patients. Abnormally low HbA2
were seen in some SCD patients without alpha thalassemia gene deletion.
Certain structural Hb variants may also reduce the level of HbA2 at varying degrees.[44] The patients with abnormally low HbA2
had higher levels of some minor components in areas designated for
HbA1a, and HbA1b, HbA1c, which are post-translational modifications of
the major haemoglobin component A0. Since HbSS individuals do not have HbA0, these may represent unidentified modified variants of HbS which compete with HbA2 for the alpha chain. The elevated HbA2
in SCD and HbAS individuals can be attributed to the combination of
alpha chain preferably with available normal delta chain over abnormal
beta S chain,[12] especially in the presence of limiting alpha chains as is the case in alpha thalassemia. The reduced HbA2
level observed in HbAA individuals with coinheritance of 2 alpha
deletion is due to the underproduction of α globin chains that could
combine with delta chains to form HbA2. Our findings corroborate previous reports that a reduction in HbA2 level is sometimes indicative of α-thalassaemia trait in individuals with normal Hb genotype,[45]
unlike in the SCD population. These HbAA individuals with alpha
thalassemia have ineffective erythropoiesis and excess beta-globin
chains, which precipitate to cause haemolysis and reduced haemoglobin
level. This may contribute to the lower HGB and HCT observed (though
not statistically significant) in our thalassemic control population.
HbF level ≥20% (HbSF) was associated with a significantly lower HbA2 level. The inverse relationship between HbF and HbA2
may be explained by the fact that the more the gamma chains are
available to combine with the alpha chains to form HbF, the fewer are
the alpha chains available to combine with the delta chains to form HbA2 and vice versa.
The sample size of patients in some of the genotype groups such as HbSSα+2,
HbSC and SF is small, and a validation of the above findings in a
larger cohort of patients with a higher number of individuals with
these less common SCD genotypes is therefore desirable.
Conclusions
Our
results have shed more light on the heterogeneity of SCD among our
patients. The study also demonstrated that C haemoglobin and homozygous
alpha-thalassemia tend to ameliorate the haematological parameters more
than the HbF level in our SCD population. Thus, the haematological
effects of alpha thalassemia on HbA2, HGB and HCT in SCD patients may be reversed in normal HbAA individuals.
References
- Waye JS & Chui DH. The alpha globin gene cluster: genetics and disorders. Clin Invest Med 2001; 24(2), 103-109.
- Milner
PF, Garbutt GJ, Nolan-Davis LV, Jonah F, Wilson LB, Wilson JT. Habara
A, Steinberg MH. Minireview: Genetic basis of heterogeneity and
severity in sickle cell disease. Exp Biol Med (Maywood).
2016;241(7):689-96. https://doi.org/10.1177/1535370216636726 PMid:26936084 PMCid:PMC4950383
- Thein SL. Genetic modifiers of sickle cell disease. Hemoglobin. 2011; 35(5-6):589-606. https://doi.org/10.3109/03630269.2011.615876 PMid:21967611
- Serjeant
GR. The Geography of Sickle Cell Disease: Opportunities for
Understanding its Diversity. Annals of Saudi Medicine, 1994;
14(3):237-246 https://doi.org/10.5144/0256-4947.1994.237 PMid:17586900
- Falusi AG & Esan GF. Foetal haemoglobin levels in sickle cell anaemia in Nigerians Afr. J Med Med Sci 1989; 18: 145-149
- Kotila
TR, Fawole OI, Shokunbi WA Haemoglobin F and clinical severity of
sickle cell anaemia among Nigerian adults Afr J Med Med Sci.
2000;29(3-4):229-31.
- Murray N, Serjeant
BE, Serjeant GR. Sickle cell-hereditary persistence of fetal
haemoglobin and its differentiation from other sickle cell syndromes.
Br J Haematol. 1988;69:89-92 https://doi.org/10.1111/j.1365-2141.1988.tb07607.x PMid:2454649
- Stevens
MCG, Hayes RJ, Vaidya S. Serjeant GR: Fetal hemoglobins and clinical
severity of homozygous sickle cell disease in early childhood. J
Pediatr 1981; 98:37-41. https://doi.org/10.1016/S0022-3476(81)80529-X
- Adeyemo
TA, Ojewunmi OO, Oyetunji IA, Rooks H, Rees DC, Akinsulie AO, et al. A
survey of genetic fetal-haemoglobin modifiers in Nigerian patients with
sickle cell anaemia. PLoS ONE 2018; 13(6): e0197927. https://doi.org/10.1371/journal.pone.0197927 PMid:29879141 PMCid:PMC5991720
- Falusi
A.G., Esan G.J.F., Ayyuo H., Higgs D.R. Alpha- thalassaemia in Nigeria:
Its interaction with Sickle Cell Disease. Eur J. Haematol
1987;38:370-375. https://doi.org/10.1111/j.1600-0609.1987.tb00013.x PMid:3609256
- Ballas
SK, Gay RN, Chehab FF. Is Hb A2 elevated in adults with
sickle-alpha-thalassemia (beta(S)/beta(S); -alpha/-alpha)? Hemoglobin.
1997;21(5):405-450 https://doi.org/10.3109/03630269708993127 PMid:9322076
- Wild
BJ and Bain B.J. Detection and quantification of normal and variant
haemoglobins: an analytical review. Ann Clin Biochem 2004; 41:355-369 https://doi.org/10.1258/0004563041731600 PMid:15333187
- Kotila
TR. Guidelines for the diagnosis of the haemoglobinopathies in Nigeria.
Ann Ib Postgrad Med. 2010 Jun;8(1):25-9. doi: 10.4314/aipm.v8i1.63954. https://doi.org/10.4314/aipm.v8i1.63954 PMid:25161471
- Lepp C. A., Bluestein B. I. Hemoglobin electrophoresis at alkaline pH on agarose gels. Clinical Chemistry. 1978;24(6):936-937. https://doi.org/10.1093/clinchem/24.6.936
- Joutovsky
A, Hadzi-Nesic J, Nardi MA. HPLC retention time as a diagnostic tool
for hemoglobin variants and hemoglobinopathies: a study of 60 000
samples in a clinical diagnostic laboratory. Clin Chem 2004;
50:1736-1747 https://doi.org/10.1373/clinchem.2004.034991 PMid:15388656
- QIAGEN®
Sample and Assay Technologies. Gentra® Puregene® Handbook for
purification of archived DNA from human whole blood, bone marrow, buffy
coat, buccal cells, body fluids, cultured cells, tissue, mouse tail,
yeast, bacteria. Third Edition June 2011
- Liu
YT, Old JM, Miles K, Fischer CA, Weatherall DJ, Clegg JB. Rapid
detection of alpha thalassemia deletions and alpha-globin gene
triplication by multiplex polymerase chain reactions. Br J Haematol
2000; 108(2):295-299. https://doi.org/10.1046/j.1365-2141.2000.01870.x PMid:10691858
- Y.
Daniel, "Haemoglobinopathy diagnostic tests: blood counts, sickle
solubility test, haemoglobin electrophoresis and high-performance
liquid chromatography" in Practical Management of Haemoglobinopathies,
I. E. Okpala, Ed., Blackwell Publishing, 2004; 10-19. https://doi.org/10.1002/9780470988398.ch2
- Inusa
BP, Daniel Y, Lawson JO, Dada J, Matthews CE, et al. (2015) Sickle Cell
Disease Screening in Northern Nigeria: The Co- Existence of Β-
Thalassemia Inheritance. Pediat Therapeut 2015;5(3):3-6.
doi:10.4172/2161-0665.1000262 https://doi.org/10.4172/2161-0665.1000262
- Akinyanju O.O. A profile of sickle cell disease in Nigeria. Ann N Y Acad Sci. 1989; 565:126-136 https://doi.org/10.1111/j.1749-6632.1989.tb24159.x PMid:2672962
- Brown
BJ, Akinkunmi BF, Fatunde OJ. Age at diagnosis of sickle cell disease
in a developing country. African Journal of Medicine and Medical
Sciences 2010; 39(3) :221-225.
- Akinbami
A, Uche E, Dosunmu A, Osikomaiya B, Adediran A, Sarah JO, Esther O,
Badiru M, Bamiro R. Haemoglobin F and A2 profiles among sickle cell
anaemia patients in Lagos State University Teaching Hospital (LASUTH),
Nigeria. Ann Trop Pathol 2018;9:26-31 https://doi.org/10.4103/atp.atp_45_17
- Adeyemo
T, Ojewunmi O, Oyetunji A. Evaluation of high performance liquid
chromatography (HPLC) pattern and prevalence of beta-thalassaemia trait
among sickle cell disease patients in Lagos, Nigeria. Pan Afr Med J.
2014;18:71. doi: 10.11604/pamj.2014.18.71.4239. https://doi.org/10.11604/pamj.2014.18.71.4239 PMid:25400838 PMCid:PMC4230225
- Mikobi
TM, Lukusa PT, Muamba JM, Rhama T. Homozygous Deletion
Alpha-Thalassemia and Hereditary Persistence of Fetal Hemoglobin, Two
Genetic Factors Predictive the Reduction of Morbidity and Mortality
During Pregnancy in Sickle Cell Patients. A Report from the Democratic
Republic of Congo. Mediterr J Hematol Infect Dis. 2019 Jul
1;11(1):e2019039. doi: 10.4084/MJHID.2019.039 https://doi.org/10.4084/mjhid.2019.039 PMid:31308915 PMCid:PMC6613621
- Ojewunmi
OO, Adeyemo TA, Oyetunji AI, Benn Y, Ekpo MG, Iwalokun BA. Association
of alpha-thalassemia and Glucose-6-Phosphate Dehydrogenase deficiency
with transcranial Doppler ultrasonography in Nigerian children with
sickle cell anemia. J Clin Lab Anal. 2021 Jun;35(6):e23802. doi:
10.1002/jcla.23802. Epub 2021 May 3 https://doi.org/10.1002/jcla.23802 PMid:33938598 PMCid:PMC8183942
- Rumaney
MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J,
Wonkam A The Co-Inheritance of Alpha-Thalassemia and Sickle Cell Anemia
Is Associated with Better Hematological Indices and Lower Consultations
Rate in Cameroonian Patients and Could Improve Their Survival Crossref
DOI link: https://doi.org/10.1371/journal.pone.0100516 PMid:24978191 PMCid:PMC4076272
- Darbari
DS, Onyekwere O, Nouraie M, Minniti CP, Luchtman-Jones L, et al.(2012)
Markers of severe vasocclusive painful episode frequency in children
and adolescents with sickle cell anemia. Journal of Pediatrics 160:
268-290 https://doi.org/10.1016/j.jpeds.2011.07.018 PMid:21890147 PMCid:PMC3258348
- Saraf
SL, Molokie RE, Nouraie M, Sable CA, Luchtman-Jones L, Ensing GJ,
Campbell AD, Rana SR, Niu XM, Machado RF, Gladwin MT, Gordeuk VR.
Differences in the clinical and genotypic presentation of sickle cell
disease around the world Paediatr Respir Rev. 2014; 15(1): 4-12.
doi:10.1016/j.prrv.2013.11.003 https://doi.org/10.1016/j.prrv.2013.11.003 PMid:24361300 PMCid:PMC3944316
- Forget BG & Bunn HF. Classification of the Disorders of Hemoglobin. Cold Spring Harb Perspect Med. 2013; 3(2): a011684 https://doi.org/10.1101/cshperspect.a011684 PMid:23378597 PMCid:PMC3552344
- Steinberg
MH & Embury SH. Alpha-thalassemia in Blacks: Genetic and Clinical
Aspects and Interactions with the Sickle Hemoglobin Gene Blood, 1986;
68(5): 985-990. https://doi.org/10.1182/blood.V68.5.985.985 PMid:3533181
- Higgs
DR, Pressley L, Clegg JB, Weatherall DJ, Serjeant GR α Thalassaemia in
Black Populations Johns Hopkins Med. J. 1980: 146:300-10
- Upasana
Joneja, Gene Gulati, Alina Dulau Florea, Jerald Gong. The Results of
Hemoglobin Variant Analysis in Patients Revealing Microcytic
Erythrocytosis on Complete Blood Count, Laboratory Medicine, Volume 49,
Issue 2, May 2018, Pages 147-153, https://doi.org/10.1093/labmed/lmx071 PMid:29346667 PMCid:PMC6251538
- Altay
C, Gravely ME, Joseph BR, Williams DF. Alpha thalassemia-2 and the
variability of hematological values in children with sickle cell
anemia. Pediatr Res I 1981;5: 1093 https://doi.org/10.1203/00006450-198108000-00004 PMid:7267182
- Antwi-Boasiako
C, Ekem I, Abdul-Rahman M, Sey F, Doku A, Dzudzor B, Dankwah GB, Otu
KH, Ahenkorah J, Aryee R. Hematological parameters in Ghanaian sickle
cell disease patients. J Blood Med. 2018 Oct 31;9:203-209. doi:
10.2147/JBM.S169872. https://doi.org/10.2147/JBM.S169872 PMid:30464671 PMCid:PMC6217132
- Embury
SH, Dozy AM, Miller J, Davis JR, Kleman KM, Preisler H, Vichinsky E,
Lande WN, Lubin BH, Kan YW, Mentzer WC. Concurrent sickle-cell anemia
and alpha-thalassemia: effect on severity of anemia. N Engl J Med.
1982;306:270-274 https://doi.org/10.1056/NEJM198202043060504 PMid:6172710
- Steinberg
MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in
sickle cell anemia: a glass half full? Blood. 2014;123(4):481-485. doi:
10.1182/blood-2013-09-528067. https://doi.org/10.1182/blood-2013-09-528067 PMid:24222332
- Steinberg
MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in
sickle cell anemia: teinberg MH and Sebastiani P. Genetic Modifiers of
Sickle Cell Disease. Am J Hematol. 2012; 87(8): 795-803 https://doi.org/10.1002/ajh.23232 PMid:22641398 PMCid:PMC4562292
- Milner
PF, Garbutt GJ, Nolan-Davis LV, Jonah F, Wilson LB, Wilson JT.The
effect of Hb F and alpha-thalassemia on the red cell indices in sickle
cell anemia. Am J Hematol. 1986;21(4):383-395 https://doi.org/10.1002/ajh.2830210407 PMid:2420172
- Salvagno
GL, Sanchis-Gomar F, Picanza A, Lippi G. Red blood cell distribution
width: A simple parameter with multiple clinical applications, Critical
Reviews in Clinical Laboratory Sciences, 2015;52(2): 86-105, DOI:
10.3109/10408363.2014.992064 https://doi.org/10.3109/10408363.2014.992064 PMid:25535770
- Velasco-Rodríguez
D, Blas C, Alonso-Domínguez J-M, Vega G, Soto C, García-Raso A,
Llamas-Sillero P. Cut-Off Values of Hematologic Parameters to Predict
the Number of Alpha Genes Deleted in Subjects with Deletional Alpha
Thalassemia. International Journal of Molecular Sciences. 2017;
18(12):2707. https://doi.org/10.3390/ijms18122707 PMid:29236053 PMCid:PMC5751308
- Old
J, Hemoglobinopathies and Thalassemias, Editor(s): David Rimoin, Reed
Pyeritz, Bruce Korf, Emery and Rimoin's Principles and Practice of
Medical Genetics, Academic Press, 2013, Chapter 71, Pages 1-44, ISBN
9780123838346, https://doi.org/10.1016/B978-0-12-383834-6.00075-6.
- Rose MS Epitaph for the M.C.H.C. Br Med J. 1971 Oct 16; 4(5780): 169.doi: 10.1136/bmj.4.5780.169 https://doi.org/10.1136/bmj.4.5780.169 PMid:5113025 PMCid:PMC1799061
- Sarma
PR. Red Cell Indices. In: Walker HK, Hall WD, Hurst JW, editors.
Clinical Methods: The History, Physical, and Laboratory Examinations.
3rd edition. Boston: Butterworths; 1990. Chapter 152. Available from: https://www.ncbi.nlm.nih.gov/books/NBK260/
- Villegas
A, González FA, Nieto JM, de la Fuente-Gonzalo F, Martínez R, Torrejón
MJ, Ropero P. Haemoglobinopathies that occur with decreased HbA2
levels: a gene mutation set involving the δ gene at a Spanish centre. J
Clin Pathol. 2017;70(1):75-80. doi: 10.1136/jclinpath-2016-203879. https://doi.org/10.1136/jclinpath-2016-203879 PMid:27387985
- Harteveld
CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May
28;5:13. doi: 10.1186/1750-1172-5-13. https://doi.org/10.1186/1750-1172-5-13 PMid:20507641 PMCid:PMC2887799
[TOP]