Cecilia
Monaldi*1, Sara De Santis1,
Manuela Mancini2, Samantha Bruno1,
Michele Cavo1,2 and Simona Soverini1.
1
Dipartimento di Medicina Specialistica, Diagnostica e Sperimentale,
Università di Bologna, Bologna, Italy.
2 IRCCS Azienda Ospedaliero-Universitaria di
Bologna, Istituto di Ematologia “Seràgnoli”, Bologna, Italy.
Correspondence to: Cecilia Monaldi, PhD student.
Institute of
Hematology “L. e A. Seràgnoli”, Via Massarenti 9, 40138 Bologna, Italy.
Tel: +390512143791. E-mail:
cecilia.monaldi2@unibo.it
Published: July 1, 2021
Received: May 26, 2021
Accepted: June 11, 2021
Mediterr J Hematol Infect Dis 2021, 13(1): e2021046 DOI
10.4084/MJHID.2021.046
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Over
the past decade, we have witnessed significant advances in the
molecular characterization of systemic mastocytosis (SM). This has
provided important information for a better understanding of the
pathogenesis of the disease but has also practically impacted the way
we diagnose and manage it. Advances in molecular testing have run in
parallel with advances in therapeutic targeting of constitutive active
KIT, the major driver of the disease. Therefore, assessing the
molecular landscape in each SM patient is essential for diagnosis,
prognosis, treatment, and therapeutic efficacy monitoring. This is
facilitated by the routine availability of novel technologies like
digital PCR and NGS. This review aims to summarize the pathogenesis of
the disease, discuss the value of molecular diagnostic testing and how
it should be performed, and provide an overview of present and future
therapeutic concepts based on fine molecular characterization of SM
patients.
|
Introduction
Mastocytosis
is a rare neoplasm characterized by the expansion and accumulation in
different organs/tissues of clonal mast cells (MCs) that can be
recognized as morphologically and immunophenotypically abnormal.[1]
The clinical presentation of mastocytosis is heterogeneous, with
various manifestations ranging from skin-limited disease (cutaneous
mastocytosis, CM) to extra-cutaneous involvement (systemic
mastocytosis, SM). CM is frequent in the pediatric age but it can
spontaneously regress during puberty.[2,3]
In contrast, SM is generally seen in adult patients and may be
associated with multiorgan dysfunction and shortened survival.[4]
The 2016 World Health Organization (WHO) defined major categories and
variants of SM. Based on histological criteria, clinical parameters,
and organ involvement, SM is divided into indolent SM (ISM), smoldering
SM (SSM), and advanced SM variants (AdvSM), including SM with an
associated clonal hematopoietic non-MC disease (SM-AHN), aggressive SM
(ASM) and MC leukemia (MCL).[5] ISM
is the most common subtype and has a relatively benign prognosis,[6] although 5 to 10% of ISM patients
progress to SSM or AdvSM.[7,8]
SSM is associated with an increased symptom burden, inferior
survival compared to ISM, and a much higher transformation rate (15-20%
for transformation to AdvSM or acute leukemia).[9]
In addition, patients with AdvSM have a reduced life expectancy.[10]
Whereas
treatment of ISM is generally palliative and primarily directed towards
preventing anaphylaxis and the control of symptoms, treatment of AdvSM
needs cytoreductive therapy to ameliorate disease‐related organ
dysfunction. For a long time, high-dose chemotherapy possibly followed
by transplant when feasible, interferon or cladribine have represented
the main options for AdvSM patients. The knowledge of the driver lesion
underlying the molecular pathogenesis of SM has ultimately enabled the
development of targeted therapies. Despite the great hurdle represented
by the rarity of AdvSM patients, the tyrosine kinase inhibitor
midostaurin has recently been approved based on the results of phase 2
clinical trial that lasted almost 10 years.[11]
Other
investigational agents, like avapritinib and ripretinib, are being
evaluated. In addition, several additional compounds with the same or
even different targets have been tested at the preclinical level.
In
this review, we summarize the molecular studies and acquisitions that
have led to a better understanding of the pathogenesis of SM, paving
the way to the development of targeted therapies. Moreover, we discuss
how the implementation of advanced molecular technologies has recently
refined the diagnostic and prognostic algorithms and how molecular
testing is acquiring a pivotal role in treatment individualization.
Finally, we also provide an overview of a series of new putative
therapeutic targets, some of whom might find a clinical translation,
primarily via repurposing of already approved agents.
Activating KIT Mutations Drive the
Pathogenesis of SM
SM is virtually
always associated with somatic gain-of-function mutations of the
proto-oncogene c-KIT,
which encodes a transmembrane protein belonging to type III tyrosine
kinase receptors. KIT is expressed by MCs, hematopoietic progenitor
cells, germ cells, melanocytes, and interstitial cells of Cajal in the
gastrointestinal tract.[12] KIT
expression is
down-regulated upon differentiation of hematopoietic progenitors into
mature cells of all lineages, except in MCs that retain high cell
surface KIT expression levels. The KIT receptor explicitly binds the
stem cell factor (SCF) cytokine, also known as the KIT ligand, steel
factor, or MC growth factor. The interaction between KIT and its ligand
leads to dimerization of the receptor and activation of its intrinsic
tyrosine kinase activity and drives a variety of downstream signal
transduction pathways.[13] The
transduction process
involves different players such as PI3 kinase, Src family kinases, the
Ras-Erk pathway, and JAK/STAT, resulting in cell proliferation,
survival, and migration. Aberrant activation of the KIT receptor,
caused by gain-of-function mutations of the gene, is a frequent event
in SM and results in uncontrolled production and proliferation of MCs.
The first identification of somatic gain-of-function mutations in
the KIT gene occurred
in the HMC-1 human MCL cell line. Two heterozygous point mutations
of KIT were revealed:
a substitution of aspartic acid with valine at residue 816 (KIT D816V) and a
substitution of valine with glycine at residue 560 (KIT
V560G). D816V and V560G are the prototypes of two distinct categories
of activating mutations, displaying different oncogenic properties and
sensitivity to tyrosine kinase inhibitors (TKIs) (Figure 1).[14]
The first can indeed be classified as an 'enzymatic pocket' mutation
since it falls in the activation loop at the entrance of the enzymatic
pocket. The activation loop serves as a sort of molecular switch for
kinase activity, and D816V turns it permanently on. Given its strategic
position with respect to the enzymatic pocket, this mutation is an
obstacle to the binding of some TKIs, like imatinib. The second, in
contrast, belongs to the wider category of 'regulatory type' mutations.
It is located in the intracellular juxta-membrane domain of the
receptor, which has a critical auto-inhibitory function; thus, it
results in constitutive kinase activation.
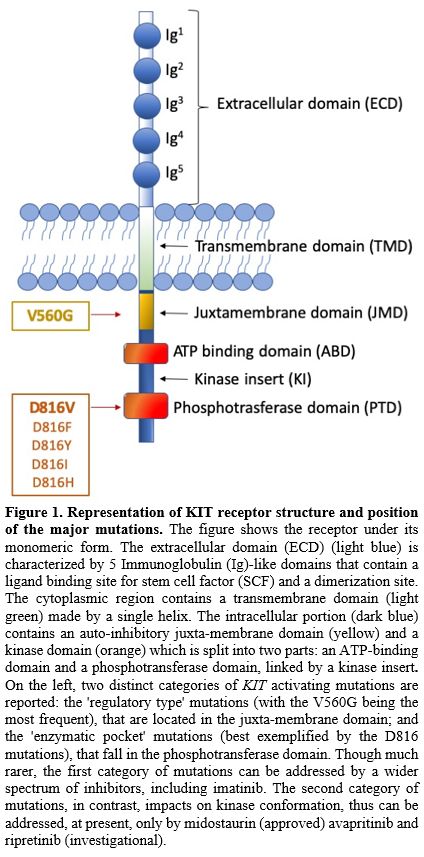 |
Figure
1. Representation of KIT
receptor structure and position of the major mutations.
The figure shows the receptor under its monomeric form. The
extracellular domain (ECD) (light blue) is characterized by 5
Immunoglobulin (Ig)-like domains that contain a ligand binding site for
stem cell factor (SCF) and a dimerization site. The cytoplasmic region
contains a transmembrane domain (light green) made by a single helix.
The intracellular portion (dark blue) contains an auto-inhibitory
juxta-membrane domain (yellow) and a kinase domain (orange) which is
split into two parts: an ATP-binding domain and a phosphotransferase
domain, linked by a kinase insert. On the left, two distinct categories
of KIT activating mutations are reported: the 'regulatory type'
mutations (with the V560G being the most frequent), that are located in
the juxta-membrane domain; and the 'enzymatic pocket' mutations (best
exemplified by the D816 mutations), that fall in the phosphotransferase
domain. Though much rarer, the first category of mutations can be
addressed by a wider spectrum of inhibitors, including imatinib. The
second category of mutations, in contrast, impacts on kinase
conformation, thus can be addressed, at present, only by midostaurin
(approved) avapritinib and ripretinib (investigational).
|
Regulatory
type' mutations may also affect the binding of substrates or signal
transducing or regulatory molecules, or induce ligand-independent
dimerization with subsequent autophosphorylation and activation of the
kinase. The greatest majority of SM patients, irrespective of the WHO
sub-type, display the D816V mutation.[4,15] More rare variants like D816Y,
D816F, D816H, and D816I have occasionally been detected. Less common
(<5%) KIT mutations at
other codons (mostly belonging to the 'regulatory type' class) have
been reported.[16,17] Rare
germline KIT
mutations associated with familial mastocytosis have also been
reported, including F522C, A533D and K509I. For these forms, the
tyrosine kinase inhibitor imatinib offers a valuable therapeutic
option.[18] Moreover, several
non-D816V mutations
have anecdotally been reported in MCL and mast cell sarcomas. Mital et
al. reported one case of MCL with a p.A502_Y503dup mutation in exon 9.[19] Other studies conducted in patients
with MCL and mast cell sarcomas described KIT mutations at
exon 10, exon 11, and exon 13.[20-22]
Finally, children with CM display missense KIT
mutations targeting exon 17 at codon 816 but also several alterations
in exons 8 and 9 involving the fifth Ig loop of the KIT extracellular
domain and leading to constitutive activation of the receptor.[23]
The
abnormal kinase activity of KIT has been documented in other human
malignancies such as germ cell tumors, melanoma, GIST, and acute
myeloid leukemia (AML).[24-27]
The Role of KIT Mutation Detection in
the Diagnostic Workout of SM
CThe
current WHO classification of mastocytosis defines one major and four
minor diagnostic criteria for SM.5 The major SM criterion is the
presence of multifocal dense aggregates of MCs in bone marrow (BM) or
other visceral organs (at least 15 MCs/cluster). Minor SM criteria
include: i) abnormal morphology of MCs (>25%), ii) expression of
CD2
and/or CD25 in MCs, iii) persistent serum tryptase concentration
>20
ng/mL, and iv) presence of the KIT
D816V point mutation in the BM or another extracutaneous organ. If at
least one major and one minor or three minor SM criteria are fulfilled,
the diagnosis of SM can be established.
The European Competence
Network on Mastocytosis (ECNM) suggests an algorithm for patients with
suspected (systemic) mastocytosis and provides essential technical
guidelines for KIT mutation detection.[28]
In The
case of suspected mastocytosis in an adult patient without skin lesions
and average or slightly increased tryptase level, the presence of KIT D816V
mutation may be investigated at first in peripheral blood (PB). If the
D816V mutation is detected in PB cells, a BM examination is
recommended.[29] In adult patients
with suspected
mastocytosis and documented skin involvement, a BM investigation is
required. In the non-availability of fresh BM aspirate sample, KIT mutational
analyses can be performed on cells detached from BM smears or a
paraffin-embedded biopsy sample (Figure
2).[30]
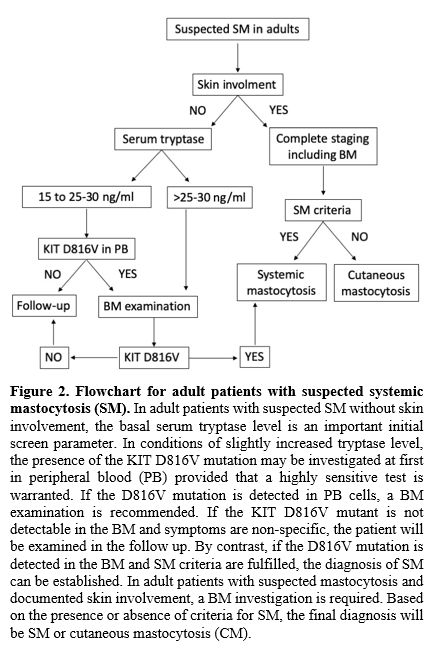 |
Figure
2. Flowchart
for adult patients with suspected systemic mastocytosis (SM).
In adult patients with suspected SM without skin involvement, the basal
serum tryptase level is an important initial screen parameter. In
conditions of slightly increased tryptase level, the presence of the
KIT D816V mutation may be investigated at first in peripheral blood
(PB) provided that a highly sensitive test is warranted. If the D816V
mutation is detected in PB cells, a BM examination is recommended. If
the KIT D816V mutant is not detectable in the BM and symptoms are
non-specific, the patient will be examined in the follow up. By
contrast, if the D816V mutation is detected in the BM and SM criteria
are fulfilled, the diagnosis of SM can be established. In adult
patients with suspected mastocytosis and documented skin involvement, a
BM investigation is required. Based on the presence or absence of
criteria for SM, the final diagnosis will be SM or cutaneous
mastocytosis (CM).
|
In
most SM patients, particularly ISM patients, the MC burden, that is,
the number of neoplastic MCs infiltrating the BM, is very low.
Moreover, mononuclear cells (MNCs) from PB are almost
invariably KIT
D816V negative when investigated by low sensitivity techniques. This
means that sensitivity is crucial to avoid false-negative results and
misdiagnosis. Over the past few years, substantial advances have been
made to develop more and more sensitive and accurate diagnostic assays.
Chronologically, the following techniques have been proposed for KIT
D816V mutation testing. They all have advantages as well as
disadvantages and differ in terms of lower detection limit, target
mutation(s), and requirement of dedicated instrumentations: i) RT-PCR
plus restriction fragment length polymorphism (RFLP), ii) nested RT-PCR
followed by denaturing high-performance liquid chromatography (D-HPLC)
of amplicons, iii) peptide nucleic acid (PNA)-mediated PCR, iv)
ASO-qPCR on DNA or cDNA, v) droplet digital PCR (ddPCR) and vi)
Next-generation sequencing (NGS).
RT-PCR
plus RFLP: it is a simple, cheap, and fast technique that
allows identifying the KIT
D816V mutation with a sensitivity of 0,05%. No dedicated
instrumentation is needed except for a thermal cycler and an
electrophoretic system. However, it is not quantitative, and it does
not allow the detection of D816 variants and the identification of
mutations at other codons.[31]
Nested
RT-PCR followed by D-HPLC of amplicons: this assay allows
detecting different KIT mutations in
the same reaction.[32]
Nevertheless, it has a relatively low sensitivity (0,5-1%), and it is
not quantitative. Moreover, it is time-consuming and needs expensive
and not widely available instrumentation (D-HLPC).
PNA-mediated
PCR: it is the recommended method for FFPE BM trephine
biopsies.[31] It allows detection
of KIT
mutations at position 816 or in adjacent codons, but it is not a
quantitative assay, and it has an intermediate sensitivity (0,1%).[33]
ASO-qPCR:
it is a simple, fast, cheap, reliable, and highly sensitive method
(0,01%) that allows to detect and quantitate KIT D816V in
different tissues and specimens (BM, PB, and organ biopsies).[34,35]
However, it requires standard curves for the quantitative assessment,
thorough validation, and possibly multi-laboratory standardization.
Droplet
Digital PCR: it is a promising new method for sensitive
and accurate quantification of KIT D816V with a
sensitivity of 0,01%.[36]
Moreover, ddPCR has been shown to sensitively and reproducibly detect
and quantify KIT D816V in FFPE
material.[37]
In contrast to ASO-qPCR, absolute quantification does not need a
standard curve; thus, it is more straightforward. Commercial kits are
available.
Next-generation
sequencing (NGS):
it has the lowest sensitivity (1-5%), but it is the best remaining
option to study those patients who have no evidence of mutations at
codon 816. Commercial myeloid panels are available that include several
KIT hotspot
exons among their target genes. Such panels may also highlight the
presence of mutations in genes other than KIT, providing additional
prognostic information (see below). Being by far the most common, the
presence of a KIT D816V needs to be investigated first. However, in the
case of D816V negativity, variants like D816Y, D816H, etc., should be
ruled out. If no mutation at all can be identified at codon 816, more
rare mutations in other KIT exons (like those mentioned in the previous
section) should be investigated. A stepwise approach is thus needed.
For D816V detection and quantitation, ASO-qPCR or ddPCR are the most
frequent option. D816 variants in confirmed D816V-negative patients may
be investigated using PNA-mediated PCR. Mutations in other hotspot
regions of the KIT
gene may then be assessed by NGS (Figure
3).
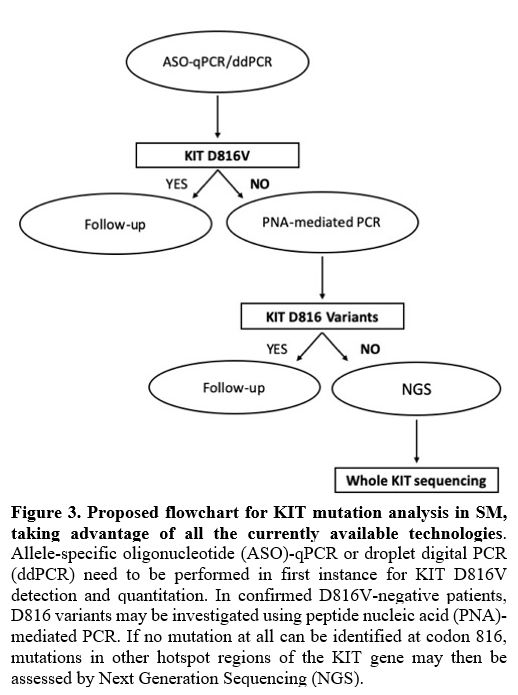 |
Figure
3. Proposed flowchart
for KIT mutation analysis in SM, taking advantage of all the currently
available technologies.
Allele-specific oligonucleotide (ASO)-qPCR or droplet digital PCR
(ddPCR) need to be performed in first instance for KIT D816V detection
and quantitation. In confirmed D816V-negative patients, D816 variants
may be investigated using peptide nucleic acid (PNA)-mediated PCR. If
no mutation at all can be identified at codon 816, mutations in other
hotspot regions of the KIT gene may then be assessed by Next Generation
Sequencing (NGS).
|
Importantly,
ASO-qPCR and ddPCR have also shown the capability to detect the D816V
mutation non-invasively in the PB.[32,37-39] Their potential for the study of
cell-free DNA is under investigation.
Prognostic Implications of KIT D816v
Allele Burden Quantification and of Multilineage Involvement
For the
assessment of KIT D816V,
quantitative methods are desirable for several reasons. The measurement
of KIT
D816V mutation percentage (the so-called 'allele burden') has
prognostic significance and can also be employed for monitoring
treatment response and disease progression. Recent studies show that
the KIT D816V allele
burden is indicative of the WHO SM subtype and correlated with the
severity of the disease.[36,40] Indeed, patients with AdvSM show a
significantly higher KIT D816V
mutation percentage compared to ISM and CM patients, the latter showing
the lowest KIT D816V allele burden. The higher KIT D816V allele
burden in AdvSM may be associated with the presence of non-MC-lineage
cells harboring the KIT
D816V mutation and could explain why it is not always correlated with
the number of MC in BM biopsies, and it is only moderately associated
with serum tryptase levels. It is now well known that some ISM and all
AdvSM patients display a multilineage disorder whose characteristics
are closely related to the risk of disease progression.[41]
In fact, the KIT
D816V mutation is detected not only in bone marrow MCs but also in
myeloid and lymphoid hematopoietic cell lineages, supporting the stem
cell origin of this disease starting from a hematopoietic precursor
with the ability for multilineage differentiation.[42-44]
The greater or smaller involvement of non-MC-lineage cells harboring
the KIT
D816V mutation reflects the risk of evolving to a more aggressive
subtype of the disease. Therefore, SM patients with
multilineage KIT D816V mutation
have been shown to have a higher probability of progression compared to
patients with MC-restricted KIT D816V.[41]
The
assessment of the KIT D816V allele burden also has a remarkable
potential in monitoring treatment response. Hoermann et al. observed
that in SM patients with stable clinical course, there were not
variations in the KIT D816V allele
burden, while advSM patients with disease progression showed a marked
increase.[40] In line with this,
Jawhar et al. used the measurement of KIT
D816V allele burden for monitoring midostaurin-treated AdvSM patients.
They showed that at month 6 of therapy, a significant reduction ≥25% of
the allele burden was the strongest on-treatment predictor for improved
survival.[45]
The KIT
D816V allele burden was also identified as a prognostic factor
regarding overall survival (OS), and to the purpose, a cut-off of ≥ or
<2% mutant alleles was used to define two distinct SM groups
with
different survival times.[36,40] The group under the cut-off level
had a significantly longer life expectancy compared to patients over
the 2% cut-off.
These findings are still under validation, but the wider implementation
of KIT
D816V allele burden assessment in clinical practice holds the potential
to improve treatment and monitoring of patients with SM.
Molecular Alterations Additional to KIT
Mutations
The somatic KIT
D816V mutation is undoubtedly the major driver of SM pathogenesis.
However, it is detectable both in patients with ISM, who have a
relatively benign prognosis and in patients with AdvSM, who are
frequently characterized by rapidly deteriorating clinical courses and
poor outcomes. Furthermore, while childhood-onset and adult-onset
mastocytosis are both associated with activating KIT
mutations, the natural history of the two conditions is quite
different. Together with experimental shreds of evidence suggesting
that KIT D816V alone is not a fully transforming oncoprotein,[46-49]
this emphasized the importance of a better understanding of SM
pathogenesis. Prognostically relevant mutations additional to KIT have now been
identified in AdvSM. These have been found to affect genes encoding for
epigenetic regulators (ASXL1,
DNMT3A, EZH2, TET2), transcription factors (RUNX1), signaling
molecules (CBL, JAK2,
KRAS, NRAS), or splicing factors (SRSF2, SF3B1, U2AF1).
These mutations are not specific for SM but are frequently detected in
myeloid neoplasm, including myelodysplastic syndromes (MDS),
myeloproliferative neoplasms (MPN), chronic myelomonocytic leukemia
(CMML), and acute myeloid leukemia (AML) where they are often
associated with poor prognosis and shorter survival.[50-52]
TET2
mutations are detected in 20-40% of AdvSM patients.[47,48]
Evidence in mice suggests that the cooperation between KIT D816V and the
loss of function of TET2 induces a
transformation to a more aggressive disease phenotype.[53]
Moreover, when TET2 mutations are
associated with DNMT3A
mutations, the prognosis of SM patients is severely impaired compared
to those with wild-type genes.[48]
Analogously to TET2,
the presence of other mutations additional to KIT may confer an adverse
prognosis compared with patients without such abnormalities. Jawhar et
al. were the first to observe that in SM-AHN, the presence and number
of mutated genes within the SRSF2/ASXL1/RUNX1
(S/A/R)
panel was associated with poor outcome and adverse clinical
characteristics.[54] In
particular, mutations in SRSF2
and ASXL1
were the most predictive adverse indicators concerning OS. As expected,
these mutations were observed at a higher frequency in ASM-AHN and
MCL-AHN and at a lower frequency in ISM-AHN. Muñoz-González et al.
suggested that, in addition to S/A/R
gene panel, somatic EZH2
gene mutations can be detected in ISM patients who show a higher risk
of disease progression.[55]
Moreover, the overall response rate (ORR) and OS were significantly
higher in patients S/A/Rneg
than in patients S/A/Rpos.
The Mayo Clinic group evaluated the impact of ASXL1 and/or CBL
mutations and the occurrence of ≥3 non‐KIT mutations on the clinical
outcome of AdvSM patients and showed that they were independently
associated with inferior OS.[56]
Moreover, based on clinical and molecular parameters (including ASXL1
mutations), Pardanani et al. proposed a "mutation-augmented" prognostic
scoring system to stratify advSM patients into 3 subgroups: low-,
intermediate- and high-risk with significantly different OS.[57] Alternative prognostic scoring
systems that include information about S/A/R profiling and
other molecular markers have been proposed, and others are still under
investigation.[58-61]
KIT as Therapeutic Target
The
clinical spectrum of this disease is highly heterogeneous. The
treatment should be individualized and related to symptoms and
diagnosis of each patient. Therapy of CM is mainly directed towards
skin lesions.[62] Anti-mediator
drug therapy for MC
activation symptoms represents the mainstay of treatment for
symptomatic CM, ISM and SSM however, it should be considered in all SM
patients.[63] The goal is to
regulate mediator
secretion, keep allergies under control, and counteract
osteopenia/osteoporosis. AdvSM is frequently associated with organ
damage, so cytoreductive therapy has long been considered the best
therapeutic strategy. For a long time, interferon[64-66]
and cladribine[67-69]
have represented the main options for AdvSM patients. However, the
introduction of tyrosine kinase inhibitors (TKIs) has revolutionized
the prognosis and the outcome of patients with AdvSM.[70]
Several TKIs, such as imatinib, nilotinib, and masitinib, have shown
activity against wild-type
KIT.
They belong to type II TKI inhibitors, so they recognize only the
inactive conformation of the receptor and stabilize it. The presence of
mutations in the activation loop (like KIT
D816V) that result in a constitutively active conformation of the
receptor renders it inaccessible to type II inhibitors. Imatinib (IM)
is currently approved by the FDA for the treatment of ASM with WT KIT or with unknown
KIT
mutational status. It demonstrated activity against certain
trans-membrane (F522C) and juxta-membrane (V560G) KIT mutants.[71-73] IM efficacy has also been
reported in a patient with familiar SM with KIT K509I mutation
in the germinal line.[18]
However, since the major of SM patients harbor the KIT D816V mutation,
it has a limited role in the treatment of this disease. Nilotinib was
evaluated in phase II multicenter study of SM patients with or without
KIT D816V mutation. However, it showed modest clinical benefit,
observed only in patients with WT KIT.[74]
Masitinib showed a great in vitro activity in ISM and SSM patients with
WT KIT or with KIT mutations outside the phospho-transferase domain,[75,76]
but it has yet to be tested in AdvSM. Type I TKIs (able to bind both
the active and the inactive form of kinases) tested in SM include
dasatinib, midostaurin, and avapritinib. Dasatinib is a multikinase
inhibitor that showed in vitro activity against both WT KIT and KIT
activation loop mutants (KIT D816V/Y/F).[77]
It was also shown to re-localize KIT D816V from the cytoplasm to the
cell surface. Interestingly, the extent of this re-localization
correlates with SM severity (AdvSM > ISM).[78]
However, given its short half-life, it showed poor in vivo
effectiveness.[79]
Midostaurin is another multikinase inhibitor; currently, the only TKI
approved for the treatment of AdvSM. The encouraging results related to
the use of midostaurin in a patient with MCL80 led to a multicenter
phase II study (CPKC412D2213) of midostaurin in 26 AdvSM patients.
Results consisted in an unprecedented overall response rate (ORR) of
69% regardless of KIT mutational status, with 38% of Major Responses
(MR)(complete resolution of at least one clinical(C)-finding such as
cytopenias, osteolysis with or without pathologic fractures,
hepatosplenomegaly and/or with impaired liver function and/or ascites,
and malabsorption).[81] Starting
from these data, a
global phase II trial (CPKC412D2201) was initiated to evaluate the
efficacy and safety of midostaurin in 89 AdvSM patients.[82]
The ORR was 60% and was independent of KIT mutation status and the
number of previous therapies. Almost half of the patients (45%) had an
MR, 15% had a partial response. After the closure of this study, a
compassionate use program of midostaurin was approved in France. ORR
was 71%, with a median response duration of 17 months.[83]
The most frequents adverse effects of midostaurin are low grade
(nausea, vomiting, diarrhea), but also new or worsening grade 3 or 4
(neutropenia, anemia, and thrombocytopenia) adverse events can be
observed.
Finally,
avapritinib is a type I kinase inhibitor that selectively inhibits the
activation-loop mutants of KIT (exon 11/17, including KIT D816V).
It is approved in the USA for PDGFRA exon 18 mutant gastrointestinal
stromal tumors (GIST), and it is also in clinical development for SM
treatment. The phase I study (Explorer; NCT02561988) in adult patients
with AdvSM has demonstrated potent antineoplastic activity of
avapritinib across all advSM subtypes with complete (disappearance of
all target lesions) and durable responses.[84]
The
ORR was 77%; 74% of patients maintained the response for at least 12
months. Avapritinib was also showed to improve overall symptoms of SM.
Avapritinib is currently being evaluated in an ongoing, double-blind,
placebo-controlled, phase II study (Pioneer), where ISM patients with
moderate or severe symptoms, refractory to ≥2 best supportive care
drugs, are enrolled.[85] Most
adverse reactions of this drug are of grade 1 or 2.
Another
agent under investigation is ripretinib (DCC-2618). It is a type II
kinase inhibitor designed to inhibit the full spectrum of mutant KIT
and PDGFRA. In addition, it shows strong activity against activation
loop mutants, previously thought to be only achievable with type I
inhibitors.[86] Its efficacy and
safety are being
tested in a Phase I open-label dose-escalation study (NCT02571036) to
treat AdvSM, GIST, and advanced cancer.
Other Non-KIT Targets and Novel Agents
Several
studies have shed light on novel altered pathways involved in the
pathogenesis of SM. Under normal conditions, SCF binds KIT receptor
leading to its dimerization and activation. This binding activates
several downstream signaling molecules involved in the proliferation
and survival of MCs, such as the phosphatidylinositol triphosphate
kinase (PI3K), the Janus kinase (JAK)/signal transducers and activators
of transcription (STAT), and the rat sarcoma (RAS)/extracellular
signal-regulated kinases (ERK). The presence of the KIT D816V mutation
contributes to divert these pathways leading to a malignant
transformation of MCs.
Baumgartner
et al. showed that the SM-related oncoprotein KIT D816V promotes STAT5
activation and phosphorylation and that pSTAT5 contributes to the
growth of neoplastic MCs.[87]
Moreover, STAT5 was found to localize to the cytoplasm and to form a
signaling complex with PI3K,[88]
whose activation has been described to be constitutively associated
with D816V KIT mutant.[89]
Once activated, PI3K activates its downstream effector AKT promoting
MCs growth and survival. This evidence suggests that the STAT5-PI3K-AKT
pathway plays an important role in the pathogenesis of SM. Indeed, the
knock-down of either STAT5 or AKT activity resulted in growth
inhibition of KIT D816V+ neoplastic MCs.[88]
Therefore, drugs targeting the STAT5- PI3K-AKT axis should be
considered a potentially novel therapeutic approach in SM patients.
Another interesting strategy may be targeting the JAK/STAT signaling
pathway. Starting from the evidence that there is often an increased
expression of JAK/STAT pathway components in SM, Lasho et al. showed
KIT D816V-mediated cell growth decrease in MC cell lines treated with
the JAK2 inhibitor TG101348.[90]
One
of the major pathways downstream PI3K is represented by rapamycin
(mTOR). It is a serine/threonine kinase found in two protein complexes
called mTOR complex 1 (mTORC1) and complex 2 (mTORC2). PI3K is able to
regulate the mTORC1 pathway by the activation of AKT. Once activated,
AKT directly phosphorylates the negative regulator of mTOR, leading to
mTOR activation and phosphorylation of two effector molecules: p70
ribosomal S6 kinase (p70S6K) and eukaryotic initiation factor
4E-binding protein1 (4E-BP1).[91]
The increased activation of the mTORC1 pathway, reported in neoplastic
MCs lines and immature MCs,[92]
seems to contribute to MCs dysregulated proliferation and survival.
Therefore, the inhibition of mTORC1 by rapamycin should be considered
to contrast this event. Indeed, it has been shown to block
mTORC1-dependent p70S6K phosphorylation.[91]
Moreover, the dual PI3K/mTOR blocker NVP-BEZ235 showed profound
inhibitory effects on the growth of primary and neoplastic MCs in vitro.[93]
In
addition to KIT downstream effectors, other non-KIT targets have been
described as therapeutically interesting. For example, MCL-1, a BCL-2
family member with antiapoptotic properties, was expressed
constitutively both in neoplastic MCs of all SM variants and MC
leukemia cell lines HMC-1.1 (D816V negative) and HMC-1.2 (D816V
positive); furthermore, MCL-1 knock-down mediated by antisense
oligonucleotides or specific siRNA resulted in increased apoptosis and
a decreased survival of neoplastic MCs, showing synergistic effects in
combination with midostaurin and other TKIs including nilotinib and
imatinib.[94] These data indicate
that MCL-1 may be a novel, potential target in neoplastic MCs.
KIT
D816V was also found to downregulate BIM expression.[95]
BIM is a proapoptotic member of the BCL-2 family that acts as a tumor
suppressor in various myeloid neoplasms.[96-98]
The KIT-targeting drug midostaurin was able to induce BIM re-expression
in neoplastic HMC-1.1 and HMC-1.2 cells and to promote growth
inhibition in both subclones.95 Moreover, in the HMC-1 cell line, the
use of the proteasome inhibitor bortezomib was associated with a
substantial increase in BIM expression and induction of apoptosis.
Indeed, several studies have shown that phosphorylated BIM degradation
in neoplastic myeloid cells is usually mediated by the proteasome.[97,99]
In addition to midostaurin and bortezomib, Aichberger et al. showed
that the pan Bcl-2 blocker obatoclax contributed to inducing growth
arrest and promoting apoptosis in HMC-1 cells and had synergic effects
with midostaurin.[95] In summary,
BIM acts as a
regulator of the growth and survival of neoplastic human MCs.
Therefore, promoting BIM re-expression or targeting antiapoptotic
members of the Bcl-2 family, combined with a KIT inhibitor, may be a
novel, interesting approach in AdvSM patients.
Finally,
the loss of function of the SETD2
tumor suppressor gene has recently been reported in AdvSM.[100] It was already observed in
various solid tumors and hematologic malignancies of both myeloid and
lymphoid origin.[101] The SETD2 tumor
suppressor gene encodes the only methyltransferase able to
tri-methylate histone H3 on lysine 36 (H3K36Me3).[102]
H3K36Me3 by SETD2 is critical for the maintenance of chromatin
structure and transcriptional fidelity. Moreover, SETD2 plays pivotal
roles in RNA alternative splicing regulation, DNA damage repair, and
cytoskeleton protein methylation.[101,103]
SETD2 loss of function may be due to biallelic missense or truncating
mutations, as seen in acute leukemias, or associated with monoallelic
deletions at chromosome 3p and mutations of the remaining allele, as
reported in clear cell renal cell carcinoma (ccRCC). However, other
mechanisms acting at the transcript or protein level can be observed.
In SM, a new post-translational mechanism of SETD2 loss of function has
been described: both subclones of the HMC-1 cell line and up to 80% of
patients with AdvSM displayed H3K36Me3 deficiency as a result of
non-genomic loss of function of SETD2, in the absence of mutations or
structural aberrations.[100] The
extent of
H3K36Me3/SETD2 downmodulation was correlated with disease
aggressiveness (AdvSM>ISM). Proteasome inhibition restored
H3K36Me3
and SETD2 protein expression, suggesting that a functional protein is
produced but rapidly degraded. Moreover, the treatment with the
proteasome inhibitor bortezomib resulted in induction of apoptosis and
reduced colony growth both in the HMC-1 cell line and in primary
neoplastic MCs from patients with AdvSM. SETD2 loss of function in
acute leukemias is often associated with chromosomal aberrations and
activated gene expression in the mTOR and Jak/Stat signaling pathways,
contributing directly to leukemogenesis in acute leukemias and SM. All
these data suggest that reverting SETD2 loss of function, in
combination with KIT-targeting drugs, could be a promising therapeutic
strategy to improve the prognosis of AdvSM patients who do not respond
to or relapse on midostaurin. Further investigations into the
mechanisms leading to SETD2 altered turnover in SM and the cooperative
effects of KIT constitutive activation and SETD2 loss of function in
AdvSM are warranted.
Conclusions and Future Perspectives
Over
the past decade, we have witnessed major advances in the molecular
characterization of SM. This has provided important information for a
better understanding of the pathogenesis of the disease but has also
practically impacted the way we diagnose and manage it.
Sensitive
and accurate assessment of KIT
mutation status is critical for appropriate diagnostic and therapy.
Moreover, quantitation of KIT
D816V
allele burden has prognostic significance and can be employed for
monitoring treatment response and disease progression. Cooperating
mutations in genes other than KIT contribute to the greater
aggressiveness of the disease and may provide additional prognostic
information. Over the past few years, substantial advances have been
made in developing more sensitive and accurate diagnostic assays that
have shown the capability to detect the KIT
D816V mutation percentage at very low levels (down to 0,001%), and
non-invasively in the PB (ASO-qPCR, ddPCR). Both these features are
important, considering that SM is generally underdiagnosed.
Moreover,
a role for targeted NGS in identifying KIT non-D816V mutants or
mutations in other key myeloid genes has been recently established. The
same myeloid panels commonly interrogated in diagnostic laboratories
for acute myeloid leukemias, myelodysplasias, Philadelphia
chromosome-negative myeloproliferative neoplasms etc are suitable,
facilitating the application of relatively expensive and
high-throughput technology to such a rare setting like SM patients.
However, molecular testing will require standardization, and although
the ECNM has already provided some useful technical recommendations in
the past[29] updated and more
detailed guidelines and greater harmonization will be needed.
The
recent approval of the pan-inhibitor midostaurin (effective against
both wild-type and mutant KIT)
for the treatment of AdvSM patients has represented a breakthrough and
has paved the way for targeted therapy of SM. Additional KIT
inhibitors, like avapritinib and ripretinib, are currently being
evaluated in clinical trials. As more and more options may become
available, comprehensive molecular profiling can assist in
individualizing therapy, including treatment intensification of ISM and
SSM cases at higher risk of progression. Other novel drug targets
(STAT5-PI3K-AKT pathway, mTORC1, MCL-1, BIM, SETD2) have been validated
at the preclinical level, but more studies would need to be undertaken
before the respective inhibitors can advance to clinical use. Patients
with AdvSM failing or not tolerating approved therapies and unfit for
transplant might indeed benefit from additional therapeutic options
chosen on the basis of their molecular characterization. In this
specific, very small setting, repurposing of compounds already tested
and approved for other conditions could be more straightforward.
Thus,
biological and clinical research move forward in close synergy in SM to
offer these rare and long-neglected patients the best quality of life
and outcome expectations.
References
- Horny HP, Sotlar K,
Valent P. Mastocytosis: state of the art. Pathobiology.
2007;74(2):121-32. https://doi.org/10.1159/000101711
PMid:17587883
- Wiechers
T, Rabenhorst A, Schick T, Preussner LM, Förster A, Valent P, Horny HP,
Sotlar K, Hartmann K. Large maculopapular cutaneous lesions are
associated with favorable outcome in childhood-onset mastocytosis. J
Allergy Clin Immunol. 2015;136(6):1581-1590. https://doi.org/10.1016/j.jaci.2015.05.034
PMid:26152315
- Hartmann K, Metcalfe DD.
Pediatric mastocytosis. Hematol Oncol Clin North Am. 2000;14(3):625-40.
https://doi.org/10.1016/S0889-8588(05)70299-9
- Lim
KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, McClure
RF, Li CY, Pardanani A. Systemic mastocytosis in 342 consecutive
adults: survival studies and prognostic factors. Blood.
2009;113(23):5727-36. https://doi.org/10.1182/blood-2009-02-205237
PMid:19363219
- Valent
P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification
and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. https://doi.org/10.1182/blood-2016-09-731893
PMid:28031180 PMCid:PMC5356454
- Pieri
L, Bonadonna P, Elena C, Papayannidis C, Grifoni FI, Rondoni M,
Girlanda S, Mauro M, Magliacane D, Elli EM, Iorno ML, Almerigogna F,
Scarfì F, Salerno R, Fanelli T, Gesullo F, Corbizi Fattori G, Bonifacio
M, Perbellini O, Artuso A, Soverini S, De Benedittis C,
Muratori
S, Pravettoni V, Cova V, Cortellini G, Ciceri F, Cortelezzi A,
Martinelli G, Triggiani M, Merante S, Vannucchi AM, Zanotti R. Clinical
presentation and management practice of systemic mastocytosis. A survey
on 460 Italian patients. Am J Hematol. 2016;91(7):692-9. https://doi.org/10.1002/ajh.24382
PMid:27060898
- Trizuljak
J, Sperr WR, Nekvindová L, Elberink HO, Gleixner KV, Gorska A, Lange M,
Hartmann K, Illerhaus A, Bonifacio M, Perkins C, Elena C, Malcovati L,
Fortina AB, Shoumariyeh K, Jawhar M, Zanotti R, Bonadonna P, Caroppo F,
Zink A, Triggiani M, Parente R, von Bubnoff N, Yavuz AS, Hägglund H,
Mattsson M, Panse J, Jäkel N, Kilbertus A, Hermine O, Arock M, Fuchs D,
Sabato V, Brockow K, Bretterklieber A, Niedoszytko M, van Anrooij B,
Reiter A, Gotlib J, Kluin-Nelemans HC, Mayer J, Doubek M, Valent P.
Clinical features and survival of patients with indolent systemic
mastocytosis defined by the updated WHO classification. Allergy.
2020;75(8):1927-1938. https://doi.org/10.1111/all.14248
PMid:32108361 PMCid:PMC7115854
- Pardanani
A, Tefferi A. Systemic mastocytosis in adults: a review on prognosis
and treatment based on 342 Mayo Clinic patients and current literature.
Curr Opin Hematol. 2010 Mar;17(2):125-32. https://doi.org/10.1097/MOH.0b013e3283366c59
PMid:20075725
- Tefferi
A, Shah S, Reichard KK, Hanson CA, Pardanani A. Smoldering
mastocytosis: Survival comparisons with indolent and aggressive
mastocytosis. Am J Hematol. 2019;94(1):E1-E2. https://doi.org/10.1002/ajh.25302
PMid:30281840
- Ustun
C, Arock M, Kluin-Nelemans HC, Reiter A, Sperr WR, George T, Horny HP,
Hartmann K, Sotlar K, Damaj G, Hermine O, Verstovsek S, Metcalfe DD,
Gotlib J, Akin C, Valent P. Advanced systemic mastocytosis: from
molecular and genetic progress to clinical practice. Haematologica.
2016;101(10):1133-1143. https://doi.org/10.3324/haematol.2016.146563
PMid:27694501
- Gilreath
JA, Tchertanov L, Deininger MW. Novel approaches to treating advanced
systemic mastocytosis. Clin Pharmacol. 2019;11:77-92. https://doi.org/10.2147/CPAA.S206615
PMid:31372066 PMCid:PMC6630092
- Miettinen
M, Lasota J. KIT (CD117): a review on expression in normal and
neoplastic tissues, and mutations and their clinicopathologic
correlation. Appl Immunohistochem Mol Morphol. 2005;13(3):205-20. https://doi.org/10.1097/01.pai.0000173054.83414.22
PMid:16082245
- Valent
P, Spanblöchl E, Sperr WR, Sillaber C, Zsebo KM, Agis H, Strobl H,
Geissler K, Bettelheim P, Lechner K. Induction of differentiation of
human mast cells from bone marrow and peripheral blood mononuclear
cells by recombinant human stem cell factor/kit-ligand in long-term
culture. Blood. 1992;80(9):2237-45. https://doi.org/10.1182/blood.V80.9.2237.2237
PMid:1384799
- Longley
BJ, Reguera MJ, Ma Y. Classes of c-KIT activating mutations: proposed
mechanisms of action and implications for disease classification and
therapy. Leuk Res. 2001 Jul;25(7):571-6. https://doi.org/10.1016/S0145-2126(01)00028-5
- Garcia-Montero
AC, Jara-Acevedo M, Teodosio C, Sanchez ML, Nunez R, Prados A,
Aldanondo I, Sanchez L, Dominguez M, Botana LM, Sanchez-Jimenez F,
Sotlar K, Almeida J, Escribano L, Orfao A. KIT mutation in mast cells
and other bone marrow hematopoietic cell lineages in systemic mast cell
disorders: a prospective study of the Spanish Network on Mastocytosis
(REMA) in a series of 113 patients. Blood 2006;108(7):2366-72. https://doi.org/10.1182/blood-2006-04-015545
PMid:16741248
- Tang
X, Boxer M, Drummond A, Ogston P, Hodgins M, Burden AD. A germline
mutation in KIT in familial diffuse cutaneous mastocytosis. J Med
Genet. 2004;41(6):e88. https://doi.org/10.1136/jmg.2003.015156
PMid:15173254 PMCid:PMC1735799
- Georgin-Lavialle S,
Lhermitte L, Dubreuil P, Chandesris MO, Hermine O, Damaj G. Mast cell
leukemia. Blood. 2013;121(8):1285-95. https://doi.org/10.1182/blood-2012-07-442400
PMid:23243287
- Zhang
LY, Smith ML, Schultheis B, Fitzgibbon J, Lister TA, Melo JV, Cross NC,
Cavenagh JD. A novel K509I mutation of KIT identified in familial
mastocytosis-in vitro and in vivo responsiveness to imatinib therapy.
Leuk Res. 2006;30(4):373-8. https://doi.org/10.1016/j.leukres.2005.08.015
PMid:16183119
- Mital
A, Piskorz A, Lewandowski K, Wasąg B, Limon J, Hellmann A. A case of
mast cell leukaemia with exon 9 KIT mutation and good response to
imatinib. Eur J Haematol. 2011;86(6):531-5. https://doi.org/10.1111/j.1600-0609.2011.01598.x
PMid:21362052
- Akin
C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of
mastocytosis associated with a transmembrane c-kit mutation and
response to imatinib. Blood. 2004;103(8):3222-5. https://doi.org/10.1182/blood-2003-11-3816
PMid:15070706
- Georgin-Lavialle
S, Aguilar C, Guieze R, Lhermitte L, Bruneau J, Fraitag S, Canioni D,
Chandesris MO, Suarez F, Grandpeix-Guyodo C, Damaj G, Barete S, Aouba
A, Fite C, Robert C, Gaulard P, Lortholary O, Tournilhac O, Dubreuil P,
Hermine O. Mast cell sarcoma: a rare and aggressive entity--report of
two cases and review of the literature. J Clin Oncol. 2013;31(6):e90-7.
https://doi.org/10.1200/JCO.2012.41.9549
PMid:23129735
- Spector
MS, Iossifov I, Kritharis A, He C, Kolitz JE, Lowe SW, Allen SL.
Mast-cell leukemia exome sequencing reveals a mutation in the IgE
mast-cell receptor β chain and KIT V654A. Leukemia. 2012;26(6):1422-5. https://doi.org/10.1038/leu.2011.354
PMid:22173243 PMCid:PMC3368985
- Bodemer
C, Hermine O, Palmérini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS,
Hadj-Rabia S, Nasca L, Georgin-Lavialle S, Cohen-Akenine A, Launay JM,
Barete S, Feger F, Arock M, Catteau B, Sans B, Stalder JF, Skowron F,
Thomas L, Lorette G, Plantin P, Bordigoni P, Lortholary O, de Prost Y,
Moussy A, Sobol H, Dubreuil P. Pediatric mastocytosis is a clonal
disease associated with D816V and other activating c-KIT mutations. J
Invest Dermatol. 2010 Mar;130(3):804-15. https://doi.org/10.1038/jid.2009.281
PMid:19865100
- Abbaspour
Babaei M, Kamalidehghan B, Saleem M, Huri HZ, Ahmadipour F. Receptor
tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in
cancer cells. Drug Des Devel Ther. 2016;10:2443-59. https://doi.org/10.2147/DDDT.S89114
PMid:27536065 PMCid:PMC4975146
- Hartmann
K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C,
Baldus SE, Heinicke T, Thiele J, Buettner R, Longley BJ. Novel germline
mutation of KIT associated with familial gastrointestinal stromal
tumors and mastocytosis. Gastroenterology. 2005;129(3):1042-6. https://doi.org/10.1053/j.gastro.2005.06.060
PMid:16143141
- Gari
M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, Vandenberghe
E, Peake I, Reilly J. c-kit proto-oncogene exon 8 in-frame deletion
plus insertion mutations in acute myeloid leukaemia. Br J Haematol.
1999;105(4):894-900. https://doi.org/10.1046/j.1365-2141.1999.01449.x
PMid:10554798
- Goemans
BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, Loonen AH,
Hählen K, Reinhardt D, Creutzig U, Kaspers GJ, Heinrich MC. Mutations
in KIT and RAS are frequent events in pediatric core-binding factor
acute myeloid leukemia. Leukemia. 2005;19(9):1536-42. https://doi.org/10.1038/sj.leu.2403870
PMid:16015387
- Valent
P, Escribano L, Broesby-Olsen S, Hartmann K, Grattan C, Brockow K,
Niedoszytko M, Nedoszytko B, Oude Elberink JN, Kristensen T,
Butterfield JH, Triggiani M, Alvarez-Twose I, Reiter A, Sperr WR,
Sotlar K, Yavuz S, Kluin-Nelemans HC, Hermine O, Radia D, van Doormaal
JJ, Gotlib J, Orfao A, Siebenhaar F, Schwartz LB, Castells M, Maurer M,
Horny HP, Akin C, Metcalfe DD, Arock M; European Competence Network on
Mastocytosis. Proposed diagnostic algorithm for patients with suspected
mastocytosis: a proposal of the European Competence Network on
Mastocytosis. Allergy. 2014;69(10):1267-74. https://doi.org/10.1111/all.12436
PMid:24836395
- Arock
M, Sotlar K, Akin C, Broesby-Olsen S, Hoermann G, Escribano L,
Kristensen TK, Kluin-Nelemans HC, Hermine O, Dubreuil P, Sperr WR,
Hartmann K, Gotlib J, Cross NC, Haferlach T, Garcia-Montero A, Orfao A,
Schwaab J, Triggiani M, Horny HP, Metcalfe DD, Reiter A, Valent P. KIT
mutation analysis in mast cell neoplasms: recommendations of the
European Competence Network on Mastocytosis. Leukemia.
2015;29(6):1223-32. https://doi.org/10.1038/leu.2015.24
PMid:25650093 PMCid:PMC4522520
- Sotlar K. c-kit
mutational analysis in paraffin material. Methods Mol Biol.
2013;999:59- 78. https://doi.org/10.1007/978-1-62703-357-2_4
PMid:23666690
- Fritsche-Polanz
R, Jordan JH, Feix A, Sperr WR, Sunder-Plassmann G, Valent P, Födinger
M. Mutation analysis of C-KIT in patients with myelodysplastic
syndromes without mastocytosis and cases of systemic mastocytosis. Br J
Haematol. 2001;113(2):357-64. https://doi.org/10.1046/j.1365-2141.2001.02783.x
PMid:11380399
- Erben
P, Schwaab J, Metzgeroth G, Horny HP, Jawhar M, Sotlar K, Fabarius A,
Teichmann M, Schneider S, Ernst T, Müller MC, Giehl M, Marx A, Hartmann
K, Hochhaus A, Hofmann WK, Cross NC, Reiter A. The KIT D816V expressed
allele burden for diagnosis and disease monitoring of systemic
mastocytosis. Ann Hematol. 2014;93(1):81-8. https://doi.org/10.1007/s00277-013-1964-1
PMid:24281161
- Sotlar
K, Escribano L, Landt O, Möhrle S, Herrero S, Torrelo A, Lass U, Horny
HP, Bültmann B. One-step detection of c-kit point mutations using
peptide nucleic acid-mediated polymerase chain reaction clamping and
hybridization probes. Am J Pathol. 2003;162(3):737-46. https://doi.org/10.1016/S0002-9440(10)63870-9
- Schumacher
JA, Elenitoba-Johnson KS, Lim MS. Detection of the c-kit D816V mutation
in systemic mastocytosis by allele-specific PCR. J Clin Pathol.
2008;61(1):109-14. https://doi.org/10.1136/jcp.2007.047928
PMid:17526803
- Kristensen
T, Broesby-Olsen S, Vestergaard H, Bindslev-Jensen C, Møller MB;
Mastocytosis Centre Odense University Hospital (MastOUH). Circulating
KIT D816V mutation-positive non-mast cells in peripheral blood are
characteristic of indolent systemic mastocytosis. Eur J Haematol.
2012;89(1):42-6. https://doi.org/10.1111/j.1600-0609.2012.01789.x
PMid:22469616
- Greiner
G, Gurbisz M, Ratzinger F, Witzeneder N, Simonitsch-Klupp I,
Mitterbauer-Hohendanner G, Mayerhofer M, Müllauer L, Sperr WR, Valent
P, Hoermann G. Digital PCR: A Sensitive and Precise Method for KIT
D816V Quantification in Mastocytosis. Clin Chem. 2018;64(3):547-555. https://doi.org/10.1373/clinchem.2017.277897
PMid:29237714 PMCid:PMC7115889
- Greiner
G, Gurbisz M, Ratzinger F, Witzeneder N, Class SV, Eisenwort G,
Simonitsch-Klupp I, Esterbauer H, Mayerhofer M, Müllauer L, Sperr WR,
Valent P, Hoermann G. Molecular quantification of tissue disease burden
is a new biomarker and independent predictor of survival in
mastocytosis. Haematologica. 2020;105(2):366-374. https://doi.org/10.3324/haematol.2019.217950
PMid:31018976 PMCid:PMC7012478
- Jara-Acevedo
M, Teodosio C, Sanchez-Muñoz L, Álvarez-Twose I, Mayado A, Caldas C,
Matito A, Morgado JM, Muñoz-González JI, Escribano L, Garcia-Montero
AC, Orfao A. Detection of the KIT D816V mutation in peripheral blood of
systemic mastocytosis: diagnostic implications. Mod Pathol.
2015;28(8):1138-49. https://doi.org/10.1038/modpathol.2015.72
PMid:26067933
- Broesby-Olsen
S, Oropeza AR, Bindslev-Jensen C, Vestergaard H, Møller MB, Siebenhaar
F, Kristensen T, Mortz CG; Mastocytosis Centre Odense University
Hospital (MastOUH); Odense Research Centre for Anaphylaxis. Recognizing
mastocytosis in patients with anaphylaxis: value of KIT D816V mutation
analysis of peripheral blood. J Allergy Clin Immunol.
2015;135(1):262-4. https://doi.org/10.1016/j.jaci.2014.06.031
PMid:25091436
- Hoermann
G, Gleixner KV, Dinu GE, Kundi M, Greiner G, Wimazal F, Hadzijusufovic
E, Mitterbauer G, Mannhalter C, Valent P, Sperr WR. The KIT D816V
allele burden predicts survival in patients with mastocytosis and
correlates with the WHO type of the disease. Allergy. 2014;69(6):810-3.
https://doi.org/10.1111/all.12409
PMid:24750133 PMCid:PMC4896381
- Escribano
L, Alvarez-Twose I, Sánchez-Muñoz L, Garcia-Montero A, Núñez R, Almeida
J, Jara-Acevedo M, Teodósio C, García-Cosío M, Bellas C, Orfao A.
Prognosis in adult indolent systemic mastocytosis: a long-term study of
the Spanish Network on Mastocytosis in a series of 145 patients. J
Allergy Clin Immunol. 2009;124(3):514-21. https://doi.org/10.1016/j.jaci.2009.05.003
PMid:19541349
- Akin
C, Kirshenbaum AS, Semere T, Worobec AS, Scott LM, Metcalfe DD.
Analysis of the surface expression of c-kit and occurrence of the c-kit
Asp816Val activating mutation in T cells, B cells, and myelomonocytic
cells in patients with mastocytosis. Exp Hematol. 2000;28(2):140-7. https://doi.org/10.1016/S0301-472X(99)00145-9
- Yavuz
AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the
involvement of a hematopoietic progenitor cell in systemic mastocytosis
from single-cell analysis of mutations in the c-kit gene. Blood. 2002
Jul 15;100(2):661-5. https://doi.org/10.1182/blood-2002-01-0203
PMid:12091362
- Kocabas
CN, Yavuz AS, Lipsky PE, Metcalfe DD, Akin C. Analysis of the lineage
relationship between mast cells and basophils using the c-kit D816V
mutation as a biologic signature. J Allergy Clin Immunol.
2005;115(6):1155-61. https://doi.org/10.1016/j.jaci.2005.02.030
PMid:15940128
- Jawhar
M, Schwaab J, Naumann N, Horny HP, Sotlar K, Haferlach T, Metzgeroth G,
Fabarius A, Valent P, Hofmann WK, Cross NCP, Meggendorfer M, Reiter A.
Response and progression on midostaurin in advanced systemic
mastocytosis: KIT D816V and other molecular markers. Blood.
2017;130(2):137-145. https://doi.org/10.1182/blood-2017-01-764423
PMid:28424161
- Schwaab
J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, Kohlmann A,
Grossmann V, Meggendorfer M, Horny HP, Valent P, Jawhar M, Teichmann M,
Metzgeroth G, Erben P, Ernst T, Hochhaus A, Haferlach T, Hofmann WK,
Cross NC, Reiter A. Comprehensive mutational profiling in advanced
systemic mastocytosis. Blood. 2013;122(14):2460-6. https://doi.org/10.1182/blood-2013-04-496448
PMid:23958953
- Tefferi
A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Finke CM,
Mullally A, Li CY, Pardanani A, Gilliland DG. Frequent TET2 mutations
in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA
correlates. Leukemia. 2009;23(5):900-4. https://doi.org/10.1038/leu.2009.37
PMid:19262599 PMCid:PMC4654631
- Traina
F, Visconte V, Jankowska AM, Makishima H, O'Keefe CL, Elson P, Han Y,
Hsieh FH, Sekeres MA, Mali RS, Kalaycio M, Lichtin AE, Advani AS, Duong
HK, Copelan E, Kapur R, Olalla Saad ST, Maciejewski JP, Tiu RV. Single
nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL
mutations are present in systemic mastocytosis. PLoS One.
2012;7(8):e43090. https://doi.org/10.1371/journal.pone.0043090
PMid:22905207 PMCid:PMC3419680
- Jawhar
M, Schwaab J, Schnittger S, Sotlar K, Horny HP, Metzgeroth G, Müller N,
Schneider S, Naumann N, Walz C, Haferlach T, Valent P, Hofmann WK,
Cross NC, Fabarius A, Reiter A. Molecular profiling of myeloid
progenitor cells in multi-mutated advanced systemic mastocytosis
identifies KIT D816V as a distinct and late event. Leukemia.
2015;29(5):1115-22. https://doi.org/10.1038/leu.2015.4
PMid:25567135
- Haferlach
T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S,
Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N,
Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU,
Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa
S. Landscape of genetic lesi ons in 944 patients with myelodysplastic
syndromes. Leukemia. 2014;28(2):241-7. https://doi.org/10.1038/leu.2013.336
PMid:24220272 PMCid:PMC3918868
- Kohlmann
A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, Dicker F,
Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T.
Next-generation sequencing technology reveals a characteristic pattern
of molecular mutations in 72.8% of chronic myelomonocytic leukemia by
detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin
Oncol. 2010;28(24):3858-65. https://doi.org/10.1200/JCO.2009.27.1361
PMid:20644105
- Jalili
M, Yaghmaie M, Ahmadvand M, Alimoghaddam K, Mousavi SA, Vaezi M,
Ghavamzadeh A. Prognostic Value of RUNX1 Mutations in AML: A
Meta-Analysis. Asian Pac J Cancer Prev. 2018;19(2):325-329.
- De
Vita S, Schneider RK, Garcia M, Wood J, Gavillet M, Ebert BL, Gerbaulet
A, Roers A, Levine RL, Mullally A, Williams DA. Loss of function of
TET2 cooperates with constitutively active KIT in murine and human
models of mastocytosis. PLoS One. 2014;9(5):e96209. https://doi.org/10.1371/journal.pone.0096209
PMid:24788138 PMCid:PMC4008566
- Jawhar
M, Schwaab J, Schnittger S, Meggendorfer M, Pfirrmann M, Sotlar K,
Horny HP, Metzgeroth G, Kluger S, Naumann N, Haferlach C, Haferlach T,
Valent P, Hofmann WK, Fabarius A, Cross NC, Reiter A. Additional
mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of
patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia.
2016;30(1):136-43. https://doi.org/10.1038/leu.2015.284
PMid:26464169
- Muñoz-González
JI, Jara-Acevedo M, Alvarez-Twose I, Merker JD, Teodosio C, Hou Y,
Henriques A, Roskin KM, Sanchez-Muñoz L, Tsai AG, Caldas C, Matito A,
Sánchez-Gallego JI, Mayado A, Dasilva-Freire N, Gotlib JR, Escribano L,
Orfao A, García-Montero AC. Impact of somatic and germline mutations on
the outcome of systemic mastocytosis. Blood Adv. 2018;2(21):2814-2828. https://doi.org/10.1182/bloodadvances.2018020628
PMid:30373888 PMCid:PMC6234367
- Pardanani
AD, Lasho TL, Finke C, Zblewski DL, Abdelrahman RA, Wassie EA, Gangat
N, Hanson CA, Ketterling RP, Tefferi A. ASXL1 and CBL mutations are
independently predictive of inferior survival in advanced systemic
mastocytosis. Br J Haematol. 2016;175(3):534-536. https://doi.org/10.1111/bjh.13865
PMid:26628266
- Pardanani
A, Lasho T, Elala Y, Wassie E, Finke C, Reichard KK, Chen D, Hanson CA,
Ketterling RP, Tefferi A. Next-generation sequencing in systemic
mastocytosis: Derivation of a mutation-augmented clinical prognostic
model for survival. Am J Hematol. 2016;91(9):888-93. https://doi.org/10.1002/ajh.24426
PMid:27214377
- Jawhar
M, Schwaab J, Álvarez-Twose I, Shoumariyeh K, Naumann N, Lübke J,
Perkins C, Muñoz-González JI, Meggendorfer M, Kennedy V, Metzgeroth G,
Fabarius A, Pfeifer D, Sotlar K, Horny HP, von Bubnoff N, Haferlach T,
Cross NCP, Hofmann WK, Sperr WR, García-Montero AC, Valent P, Gotlib J,
Orfao A, Reiter A. MARS: Mutation-Adjusted Risk Score for Advanced
Systemic Mastocytosis. J Clin Oncol. 2019;37(31):2846-2856. https://doi.org/10.1200/JCO.19.00640
PMid:31509472 PMCid:PMC6823885
- Muñoz-González
JI, Álvarez-Twose I, Jara-Acevedo M, Henriques A, Viñas E, Prieto C,
Sánchez-Muñoz L, Caldas C, Mayado A, Matito A, Dasilva-Freire N, Orfao
A, García-Montero AC. Frequency and prognostic impact of KIT and other
genetic variants in indolent systemic mastocytosis. Blood.
2019;134(5):456-468. https://doi.org/10.1182/blood.2018886507
PMid:31151985
- Pardanani
A, Shah S, Mannelli F, Elala YC, Guglielmelli P, Lasho TL, Patnaik MM,
Gangat N, Ketterling RP, Reichard KK, Hanson CA, Vannucchi AM, Tefferi
A. Mayo alliance prognostic system for mastocytosis: clinical and
hybrid clinical-molecular models. Blood Adv. 2018;2(21):2964-2972. https://doi.org/10.1182/bloodadvances.2018026245
PMid:30413432 PMCid:PMC6234360
- Sperr
WR, Kundi M, Alvarez-Twose I, van Anrooij B, Oude Elberink JNG, Gorska
A, Niedoszytko M, Gleixner KV, Hadzijusufovic E, Zanotti R, Bonadonna
P, Bonifacio M, Perkins C, Illerhaus A, Elena C, Merante S, Shoumariyeh
K, von Bubnoff N, Parente R, Jawhar M, Belloni Fortina A, Caroppo F,
Brockow K, Zink A, Fuchs D, Kilbertus AJ, Yavuz AS, Doubek M, Hägglund
H, Panse J, Sabato V, Bretterklieber A, Niederwieser D, Breynaert C,
Hartmann K, Triggiani M, Nedoszytko B, Reiter A, Orfao A, Hermine O,
Gotlib J, Arock M, Kluin-Nelemans HC, Valent P. International
prognostic scoring system for mastocytosis (IPSM): a retrospective
cohort study. Lancet Haematol. 2019;6(12):e638-e649. https://doi.org/10.1016/S2352-3026(19)30166-8
- Scherber RM, Borate U.
How we diagnose and treat systemic mastocytosis in adults. Br J
Haematol. 2018;180(1):11-23. https://doi.org/10.1111/bjh.14967
PMid:29048112
- Pardanani
A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk
stratification and management. Am J Hematol. 2019;94(3):363-377. https://doi.org/10.1002/ajh.25371
PMid:30536695
- Butterfield JH.
Response of severe systemic mastocytosis to interferon alpha. Br J
Dermatol. 1998;138(3):489-95. https://doi.org/10.1046/j.1365-2133.1998.02131.x
PMid:9580806
- Butterfield
JH, Tefferi A, Kozuh GF. Successful treatment of systemic mastocytosis
with high-dose interferon-alfa: long-term follow-up of a case. Leuk
Res. 2005;29(2):131-4. https://doi.org/10.1016/j.leukres.2004.05.003
PMid:15607359
- Hauswirth
AW, Simonitsch-Klupp I, Uffmann M, Koller E, Sperr WR, Lechner K,
Valent P. Response to therapy with interferon alpha-2b and prednisolone
in aggressive systemic mastocytosis: report of five cases and review of
the literature. Leuk Res. 2004;28(3):249-57. https://doi.org/10.1016/S0145-2126(03)00259-5
- Pardanani
A, Hoffbrand AV, Butterfield JH, Tefferi A. Treatment of systemic mast
cell disease with 2-chlorodeoxyadenosine. Leuk Res. 2004;28(2):127-31. https://doi.org/10.1016/S0145-2126(03)00185-1
- Böhm
A, Sonneck K, Gleixner KV, Schuch K, Pickl WF, Blatt K, Peter B,
Herrmann H, Schernthaner GH, Pehamberger H, Rabitsch W, Sperr WR,
Valent P. In vitro and in vivo growth-inhibitory effects of cladribine
on neoplastic mast cells exhibiting the imatinib-resistant KIT mutation
D816V. Exp Hematol. 2010;38(9):744-55. https://doi.org/10.1016/j.exphem.2010.05.006
PMid:20553795
- Kluin-Nelemans
HC, Oldhoff JM, Van Doormaal JJ, Van 't Wout JW, Verhoef G, Gerrits WB,
van Dobbenburgh OA, Pasmans SG, Fijnheer R. Cladribine therapy for
systemic mastocytosis. Blood. 2003;102(13):4270-6. https://doi.org/10.1182/blood-2003-05-1699
PMid:12933573
- Gilreath
JA, Tchertanov L, Deininger MW. Novel approaches to treating advanced
systemic mastocytosis. Clin Pharmacol. 2019;11:77-92. https://doi.org/10.2147/CPAA.S206615
PMid:31372066 PMCid:PMC6630092
- Akin
C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of
mastocytosis associated with a transmembrane c-kit mutation and
response to imatinib. Blood. 2004;103(8):3222-5. https://doi.org/10.1182/blood-2003-11-3816
PMid:15070706
- Zermati
Y, De Sepulveda P, Féger F, Létard S, Kersual J, Castéran N, Gorochov
G, Dy M, Ribadeau Dumas A, Dorgham K, Parizot C, Bieche Y, Vidaud M,
Lortholary O, Arock M, Hermine O, Dubreuil P. Effect of tyrosine kinase
inhibitor STI571 on the kinase activity of wild-type and various
mutated c-kit receptors found in mast cell neoplasms. Oncogene.
2003;22(5):660-4. https://doi.org/10.1038/sj.onc.1206120
PMid:12569358
- Ma
Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, McMahon
G, Longley BJ. The c-KIT mutation causing human mastocytosis is
resistant to STI571 and other KIT kinase inhibitors; kinases with
enzymatic site mutations show different inhibitor sensitivity profiles
than wild-type kinases and those with regulatory-type mutations. Blood.
2002;99(5):1741-4. https://doi.org/10.1182/blood.V99.5.1741
PMid:11861291
- Hochhaus
A, Baccarani M, Giles FJ, le Coutre PD, Müller MC, Reiter A,
Santanastasio H, Leung M, Novick S, Kantarjian HM. Nilotinib in
patients with systemic mastocytosis: analysis of the phase 2,
open-label, single-arm nilotinib registration study. J Cancer Res Clin
Oncol. 2015;141(11):2047-60. https://doi.org/10.1007/s00432-015-1988-0
PMid:26002753 PMCid:PMC4768228
- Dubreuil
P, Letard S, Ciufolini M, Gros L, Humbert M, Castéran N, Borge L, Hajem
B, Lermet A, Sippl W, Voisset E, Arock M, Auclair C, Leventhal PS,
Mansfield CD, Moussy A, Hermine O. Masitinib (AB1010), a potent and
selective tyrosine kinase inhibitor targeting KIT. PLoS One.
2009;4(9):e7258. https://doi.org/10.1371/journal.pone.0007258
PMid:19789626 PMCid:PMC2746281
- Lortholary
O, Chandesris MO, Bulai Livideanu C, Paul C, Guillet G, Jassem E,
Niedoszytko M, Barete S, Verstovsek S, Grattan C, Damaj G, Canioni D,
Fraitag S, Lhermitte L, Georgin Lavialle S, Frenzel L, Afrin LB,
Hanssens K, Agopian J, Gaillard R, Kinet JP, Auclair C, Mansfield C,
Moussy A, Dubreuil P, Hermine O. Masitinib for treatment of severely
symptomatic indolent systemic mastocytosis: a randomised,
placebo-controlled, phase 3 study. Lancet. 2017;389(10069):612-620. https://doi.org/10.1016/S0140-6736(16)31403-9
- Schittenhelm
MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C,
Deininger MW, Druker BJ, Heinrich MC. Dasatinib (BMS-354825), a dual
SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type,
juxtamembrane, and activation loop mutant KIT isoforms associated with
human malignancies. Cancer Res. 2006;66(1):473-81. https://doi.org/10.1158/0008-5472.CAN-05-2050
PMid:16397263
- Bougherara
H, Georgin-Lavialle S, Damaj G, Launay JM, Lhermitte L, Auclair C,
Arock M, Dubreuil P, Hermine O, Poul MA. Relocalization of KIT D816V to
cell surface after dasatinib treatment: potential clinical
implications. Clin Lymphoma Myeloma Leuk. 2013;13(1):62-9. https://doi.org/10.1016/j.clml.2012.08.004
PMid:23127495
- Verstovsek
S, Tefferi A, Cortes J, O'Brien S, Garcia-Manero G, Pardanani A, Akin
C, Faderl S, Manshouri T, Thomas D, Kantarjian H. Phase II study of
dasatinib in Philadelphia chromosome-negative acute and chronic myeloid
diseases, including systemic mastocytosis. Clin Cancer Res.
2008;14(12):3906-15. https://doi.org/10.1158/1078-0432.CCR-08-0366
PMid:18559612 PMCid:PMC5018899
- Gotlib
J, Berubé C, Growney JD, Chen CC, George TI, Williams C, Kajiguchi T,
Ruan J, Lilleberg SL, Durocher JA, Lichy JH, Wang Y, Cohen PS, Arber
DA, Heinrich MC, Neckers L, Galli SJ, Gilliland DG, Coutré SE. Activity
of the tyrosine kinase inhibitor PKC412 in a patient with mast cell
leukemia with the D816V KIT mutation. Blood. 2005;106(8):2865-70. https://doi.org/10.1182/blood-2005-04-1568
PMid:15972446 PMCid:PMC1895309
- DeAngelo
DJ, George TI, Linder A, Langford C, Perkins C, Ma J, Westervelt P,
Merker JD, Berube C, Coutre S, Liedtke M, Medeiros B, Sternberg D,
Dutreix C, Ruffie PA, Corless C, Graubert TJ, Gotlib J. Efficacy and
safety of midostaurin in patients with advanced systemic mastocytosis:
10-year median follow-up of a phase II trial. Leukemia.
2018;32(2):470-478. https://doi.org/10.1038/leu.2017.234
PMid:28744009
- Gotlib
J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, Awan FT,
Hexner E, Mauro MJ, Sternberg DW, Villeneuve M, Huntsman Labed A,
Stanek EJ, Hartmann K, Horny HP, Valent P, Reiter A. Efficacy and
Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med.
2016;374(26):2530-41. https://doi.org/10.1056/NEJMoa1513098
PMid:27355533
- Chandesris
MO, Damaj G, Canioni D, Brouzes C, Lhermitte L, Hanssens K, Frenzel L,
Cherquaoui Z, Durieu I, Durupt S, Gyan E, Beyne-Rauzy O, Launay D,
Faure C, Hamidou M, Besnard S, Diouf M, Schiffmann A, Niault M, Jeandel
PY, Ranta D, Gressin R, Chantepie S, Barete S, Dubreuil P, Bourget P,
Lortholary O, Hermine O; CEREMAST Study Group. Midostaurin in Advanced
Systemic Mastocytosis. N Engl J Med. 2016;374(26):2605-7. https://doi.org/10.1056/NEJMc1515403
PMid:27355555
- Radia
D, Deininger MW, Gotlib J, Bose P, Drummond MW, Hexner E , Robinson W,
Quiery A, Winton E, George T, Horny HP, Oren R , Shi H, Schmidt-Kittler
O, Mar B , DeAngelo D. Avapritinib, a potent and selective inhibitor of
Kit D816V, induces complete and durable responses in patients (pts)
with advanced systemic mastocytosis (AdvSM) (abstract no. S830 and oral
presentation). HemaSphere. 2019. https://doi.org/10.1097/01.HS9.0000561600.89121.38
- Maurer
M, Elberink HO, Gotlib J, Sabato V, Hartmann K, Broesby-Olsen S,
Castells M, Deininger M, Heaney M, George T, Siebenhaar F, Radia D,
Triggiani M, Daele P, DeAngelo D, Schmidt-Kittler O, Lin H, Morrison A,
Mar B, Maurer M. Results from PIONEER: a randomized, double-blind,
placebo-controlled, phase 2 study of avapritinib in patients with
indolent systemic mastocytosis (ISM). Oncol Res Treat. 2020;43:77-77.
- Smith
BD, Kaufman MD, Lu WP, Gupta A, Leary CB, Wise SC, Rutkoski TJ, Ahn YM,
Al-Ani G, Bulfer SL, Caldwell TM, Chun L, Ensinger CL, Hood MM,
McKinley A, Patt WC, Ruiz-Soto R, Su Y, Telikepalli H, Town A, Turner
BA, Vogeti L, Vogeti S, Yates K, Janku F, Abdul Razak AR, Rosen O,
Heinrich MC, Flynn DL. Ripretinib (DCC-2618) Is a Switch Control Kinase
Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and
PDGFRA Variants. Cancer Cell. 2019;35(5):738-751.e9. https://doi.org/10.1016/j.ccell.2019.04.006
PMid:31085175
- Baumgartner
C, Cerny-Reiterer S, Sonneck K, Mayerhofer M, Gleixner KV, Fritz R,
Kerenyi M, Boudot C, Gouilleux F, Kornfeld JW, Sillaber C, Moriggl R,
Valent P. Expression of activated STAT5 in neoplastic mast cells in
systemic mastocytosis: subcellular distribution and role of the
transforming oncoprotein KIT D816V. Am J Pathol. 2009;175(6):2416-29. https://doi.org/10.2353/ajpath.2009.080953
PMid:19893034 PMCid:PMC2789628
- Harir
N, Boudot C, Friedbichler K, Sonneck K, Kondo R, Martin-Lannerée S,
Kenner L, Kerenyi M, Yahiaoui S, Gouilleux-Gruart V, Gondry J, Bénit L,
Dusanter-Fourt I, Lassoued K, Valent P, Moriggl R, Gouilleux F.
Oncogenic Kit controls neoplastic mast cell growth through a
Stat5/PI3-kinase signaling cascade. Blood. 2008;112(6):2463-73. https://doi.org/10.1182/blood-2007-09-115477
PMid:18579792 PMCid:PMC2532813
- Sundström
M, Vliagoftis H, Karlberg P, Butterfield JH, Nilsson K, Metcalfe DD,
Nilsson G. Functional and phenotypic studies of two variants of a human
mast cell line with a distinct set of mutations in the c-kit
proto-oncogene. Immunology. 2003;108(1):89-97. https://doi.org/10.1046/j.1365-2567.2003.01559.x
PMid:12519307 PMCid:PMC1782858
- Lasho
T, Tefferi A, Pardanani A. Inhibition of JAK-STAT signaling by
TG101348: a novel mechanism for inhibition of KITD816V-dependent growth
in mast cell leukemia cells. Leukemia. 2010;24(7):1378-80. https://doi.org/10.1038/leu.2010.109
PMid:20485374
- Kim
MS, Kuehn HS, Metcalfe DD, Gilfillan AM. Activation and function of the
mTORC1 pathway in mast cells. J Immunol. 2008;180(7):4586-95. https://doi.org/10.4049/jimmunol.180.7.4586
PMid:18354181 PMCid:PMC2698706
- Smrz
D, Kim MS, Zhang S, Mock BA, Smrzová S, DuBois W, Simakova O, Maric I,
Wilson TM, Metcalfe DD, Gilfillan AM. mTORC1 and mTORC2 differentially
regulate homeostasis of neoplastic and non-neoplastic human mast cells.
Blood. 2011;118(26):6803-13. https://doi.org/10.1182/blood-2011-06-359984
PMid:22053105 PMCid:PMC3245204
- Blatt
K, Herrmann H, Mirkina I, Hadzijusufovic E, Peter B, Strommer S,
Hoermann G, Mayerhofer M, Hoetzenecker K, Klepetko W, Ghanim V, Marth
K, Füreder T, Wacheck V, Valenta R, Valent P. The
PI3-kinase/mTOR-targeting drug NVP-BEZ235 inhibits growth and
IgE-dependent activation of human mast cells and basophils. PLoS One.
2012;7(1):e29925. https://doi.org/10.1371/journal.pone.0029925
PMid:22299028 PMCid:PMC3267720
- Aichberger
KJ, Mayerhofer M, Gleixner KV, Krauth MT, Gruze A, Pickl WF, Wacheck V,
Selzer E, Müllauer L, Agis H, Sillaber C, Valent P. Identification of
MCL1 as a novel target in neoplastic mast cells in systemic
mastocytosis: inhibition of mast cell survival by MCL1 antisense
oligonucleotides and synergism with PKC412. Blood. 2007;109(7):3031-41.
https://doi.org/10.1182/blood-2006-07-032714
PMid:17110460
- Aichberger
KJ, Gleixner KV, Mirkina I, Cerny-Reiterer S, Peter B, Ferenc V,
Kneidinger M, Baumgartner C, Mayerhofer M, Gruze A, Pickl WF, Sillaber
C, Valent P. Identification of proapoptotic Bim as a tumor suppressor
in neoplastic mast cells: role of KIT D816V and effects of various
targeted drugs. Blood. 2009;114(26):5342-51. https://doi.org/10.1182/blood-2008-08-175190
PMid:19850739
- Shinjyo
T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A, Houghton PJ,
Look AT, Ozawa K, Inaba T. Downregulation of Bim, a proapoptotic
relative of Bcl-2, is a pivotal step in cytokine-initiated survival
signaling in murine hematopoietic progenitors. Mol Cell Biol.
2001;21(3):854-64. https://doi.org/10.1128/MCB.21.3.854-864.2001
PMid:11154272 PMCid:PMC86676
- Luciano
F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P.
Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its
degradation via the proteasome pathway and regulates its proapoptotic
function. Oncogene. 2003;22(43):6785-93. https://doi.org/10.1038/sj.onc.1206792
PMid:14555991
- Kuribara
R, Honda H, Matsui H, Shinjyo T, Inukai T, Sugita K, Nakazawa S, Hirai
H, Ozawa K, Inaba T. Roles of Bim in apoptosis of normal and
Bcr-Abl-expressing hematopoietic progenitors. Mol Cell Biol.
2004;24(14):6172-83. https://doi.org/10.1128/MCB.24.14.6172-6183.2004
PMid:15226421 PMCid:PMC434248
- Aichberger
KJ, Mayerhofer M, Krauth MT, Vales A, Kondo R, Derdak S, Pickl WF,
Selzer E, Deininger M, Druker BJ, Sillaber C, Esterbauer H, Valent P.
Low-level expression of proapoptotic Bcl-2-interacting mediator in
leukemic cells in patients with chronic myeloid leukemia: role of
BCR/ABL, characterization of underlying signaling pathways, and
reexpression by novel pharmacologic compounds. Cancer Res.
2005;65(20):9436-44. https://doi.org/10.1158/0008-5472.CAN-05-0972
PMid:16230407
- Martinelli
G, Mancini M, De Benedittis C, Rondoni M, Papayannidis C, Manfrini M,
Meggendorfer M, Calogero R, Guadagnuolo V, Fontana MC, Bavaro L,
Padella A, Zago E, Pagano L, Zanotti R, Scaffidi L, Specchia G, Albano
F, Merante S, Elena C, Savini P, Gangemi D, Tosi P, Ciceri F, Poletti
G, Riccioni L, Morigi F, Delledonne M, Haferlach T, Cavo M, Valent P,
Soverini S. SETD2 and histone H3 lysine 36 methylation deficiency in
advanced systemic mastocytosis. Leukemia. 2018;32(1):139-148. https://doi.org/10.1038/leu.2017.183
PMid:28663576 PMCid:PMC5770597
- Li
J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K. SETD2: an
epigenetic modifier with tumor suppressor functionality. Oncotarget.
2016;7(31):50719-50734. https://doi.org/10.18632/oncotarget.9368
PMid:27191891 PMCid:PMC5226616
- Edmunds
JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during
gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J.
2008;27(2):406-20. https://doi.org/10.1038/sj.emboj.7601967
PMid:18157086 PMCid:PMC2168397
- Park
IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, Chiang YC, Davis
IJ, Fahey CC, Hacker KE, Verhey KJ, Bedford MT, Jonasch E, Rathmell WK,
Walker CL. Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell.
2016;166(4):950-962. https://doi.org/10.1016/j.cell.2016.07.005
PMid:27518565 PMCid:PMC5101839
[TOP]