Ilaria Lazzareschi1,2, Elena Rossi3,2, Antonietta Curatola1, Giovanna Capozio1, Luca Benacquista1, Ludovica Iezzi1 and Donato Rigante1,2.
1
Department of Life Sciences and Global Health, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
2 Università Cattolica Sacro Cuore, Rome, Italy.
3
Diagnostic Imaging, Oncological Radiotherapy and Hematology Department,
Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
Correspondence to:
Donato Rigante, MD, Department of Life Sciences and Global Health,
Fondazione Policlinico Universitario A. Gemelli IRCCS, Università
Cattolica Sacro Cuore, Rome, Largo A. Gemelli 8, 00168 Rome, Italy.
Tel: +39 06 30155210. Fax: +39 06 3383211. E-mail:
donato.rigante@unicatt.it
Published: January 1, 2022
Received: September 25, 2021
Accepted: December 12, 2021
Mediterr J Hematol Infect Dis 2022, 14(1): e2022008 DOI
10.4084/MJHID.2022.008
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
A
disparate group of rare hematological diseases characterized by
impaired maturation of neutrophil granulocytes defines congenital
neutropenias. Neutropenic patients are prone to recurrent infections
beginning in the first months of life. Of interest is "cyclic
neutropenia", an ultra-rare disorder revealed by sinusoidal variations
in the neutrophil count and recurring infections every 21 days.
Diagnosis of these disorders is frequently obscured by the multiple
causes of recurrent fevers in children. The aim of this overview is to
outline the physical assessment of children presenting with early-onset
symptomatic neutropenia, identify the disease between the many medical
conditions and even emergencies which should enter in differential
diagnosis, hint at the potential management with granulocyte-colony
stimulating factor, define the risk of evolution to hematologic
malignancy, and summarize inter-professional team strategies for
improving care coordination and outcomes of patients.
|
Introduction
Neutrophil
granulocytes are the primary mediators of host innate immunity against
bacterial pathogens, and genetically determined neutrophil disorders
confer a predisposition to infections. Failure in either neutrophil
number or neutrophil function can be disclosed by the occurrence of
infections in several clinical settings. The most common etiologies of
neutropenia, defined as a reduction of the absolute number of
circulating neutrophils, are acquired and include sequestration, viral
infections, chemotherapy, and drug reactions.[1] The inherited
etiologies of neutropenia are much less common, though often more
severe.[2] Congenital neutropenias are rare disorders of myelopoiesis
characterized by impaired neutrophil differentiation with maturation
arrest at the promyelocyte stage.[3] A congenital neutropenia consists
of "statically" low neutrophil counts and is largely observed in the
first infancy, while cyclic neutropenia is defined by regularly
"cyclic" episodes of neutropenia which recur every three weeks in
toddlers.[4]
The decrease of neutrophils in both bone marrow and bloodstream can reach a nadir below 200 neutrophils/mm3
during febrile neutropenic days.[5] This recurrence of fever flares in
children might have dramatic consequences on the overall quality of
life of patients and their caregivers, displaying a negative
interference with school attendance or daily activities, and might
generate a deep sense of frustration in the families.[6] A
significantly increased risk of myelodysplastic syndrome and acute
myeloid leukemia has been reported for severe congenital neutropenia,
but treatment options are limited and no reliable tools predict this
kind of progression. Moreover, the delay to a definite diagnosis may be
of several months or even years, and children are frequently exposed to
redundant and unnecessary diagnostic procedures. Hence, the importance
of considering and properly identifying congenital neutropenia warrants
the updated overview herein presented.
The Entwined Molecular Mechanisms of Neutropenia
Congenital
neutropenia encompasses a family of disorders characterized by
neutropenia, either permanent or intermittent, both mild and severe
when the neutrophil count is below 500/mm3:
the inner working of neutropenia has been extensively investigated, and
different pathways that control programmed cell death in neutrophils
have been studied. The exact physiological basis of neutropenia remains
unclear, though many authors have confirmed an interrupted cell
production in the bone marrow.[7] There is a continuum between
permanent neutropenia versus intermittent neutropenia and different
mutations at the locus 19p13.3 of the gene encoding the enzyme
neutrophil elastase, referred to as ELANE (or ELA2), have been disclosed in most cases of congenital neutropenia.[8] The molecular machinery by which ELANE
mutations disrupt myelopoiesis is unknown. However, we know that
neutrophil elastase, a 238-amino acid protein with broad proteolytic
activities, is packaged into azurophil granules within neutrophils as a
fully active enzyme after being processed in the Golgi apparatus.[9]
The regular oscillation of white blood cell counts in cyclic
neutropenia is attributed to the excessive cell turnover in the early
neutrophil compartments and to a potential autoregulatory loop with an
inverse correlation between circulating cells and humoral regulators of
the neutrophil balance.[10] Mir et al. analyzed the subpopulations of
bone marrow cells at both peak and nadir of the neutrophil cycle in
patients with cyclic neutropenia, detecting a higher proportion of
hematopoietic stem cells at the nadir, as opposed to the peak. In
particular, they found that mRNA expression levels of ELANE
and unfolded protein response-related genes were elevated at the nadir,
differently from anti-apoptotic genes, which were reduced, and
hypothesized that some stem cells escaped the unfolded protein
response-stress: these escaper cells responded to granulocyte
colony-stimulating factor (G-CSF), generating either neutrophils
between nadir and peak or new progenitor cells during the cycle.[11]
However, the exact mechanisms governing the clock-like timing of
hematopoiesis and unfolded protein response remain to decipher and are
a matter of ongoing research.
Furthermore, accelerated apoptosis
with cell cycle arrest in the G0/G1 phase has been described in
promyelocytes of patients with severe congenital neutropenia: this can
result from misfolded elastase proteins and subsequent activation of
the unfolded protein response. In these patients ELANE
mutations lead to the mislocalization and accumulation of unfolded
proteins, creating endoplasmic reticulum stress, which affects the
survival and differentiation of granulocytes.[12] More specifically,
CD34+CD45+ hematopoietic progenitor cells deriving from pluripotent
stem cell lines of patients with congenital neutropenia display
elevated levels of reactive oxygen species and a high number of
promyelocyte leukemia protein nuclear bodies, which are hallmarks of
acute oxidative stress.[13] In general terms, by the perturbation of
mitochondrial energy metabolism, uncontrolled vesicle trafficking, or
unbalanced oxidative stress, the disease causes a maturation arrest in
myeloid precursor cells, reducing the number of circulating neutrophils
and making patients vulnerable to recurrent infections.
The Spyglass of Genotype Studies in Congenital Neutropenias
Severe
congenital neutropenia is a bone marrow failure syndrome characterized
by neutropenia present from birth, leading to frequent infections of
different severity. ELANE-related
neutropenia includes severe congenital neutropenia (also known as
Kostmann syndrome) and cyclic neutropenia, which are primary disorders
characterized by similar phenotype: recurrent fevers, skin, and
oropharyngeal inflammation. ELANE
mutations have been found in 80-to-100% of cases with cyclic
neutropenia and in 35-to-63% of cases with severe congenital
neutropenia.[14] While ELANE
mutations have been proved as the nearly-exclusive cause of cyclic
neutropenia, several mutations in other genes can explain the
pathogenesis of Kostmann syndrome. Autosomal dominant gain-of-function ELANE
mutations transmit cyclic neutropenia, but sporadic cases may arise
from new germline mutations. Kostmann syndrome, first described by the
Swedish pediatrician Rolf Kostmann, who coined the term 'infantile
genetic agranulocytosis' in 1950, is a primary immunodeficiency
associated with increased apoptosis of myeloid cells and includes
different disorders caused by protean genetic abnormalities: the
mutated genes encompass the one encoding neutrophil elastase, but also
the proteins HAX1, G6PC3, WAS and GFI1.[15]
The distinction
between congenital neutropenia and cyclic neutropenia is primarily
based on clinical findings and secondly on a molecular approach,
including single-gene testing or multigene panels. Patients' siblings
or other at-risk relatives should be evaluated by ELANE genetic testing. In general terms, ELANE
pathogenic variants include missense and nonsense variants, small
deletions or insertions in exons, splicing defects, and changes in the ELANE regulatory region.[16] Genotype-phenotype correlations have been roughly defined for ELANE-related
neutropenias. Although the patterns of pathogenic variants in
congenital neutropenia and cyclic neutropenia are distinct on a
population basis, these variants might overlap, indicating that the
distinction between the two conditions should remain clinical and only
later based on the genotype analysis.[17] Some pathogenic ELANE
variants have been associated with an overall good prognosis, and some
of these appear to be solely associated with cyclic neutropenia, having
a minimal risk of hematologic malignancies.[18]
Some patients
with severe congenital neutropenia can have homozygous or compound
heterozygous mutations in the HAX1 gene, coding for the
HCLS1-associated protein X-1 or HAX1, an ubiquitously expressed
multifunctional protein predominantly localized in mitochondria. Klein
et al. showed that HAX1 is critical for maintaining the inner
mitochondrial membrane potential and protecting myeloid cells from
apoptosis, suggesting that this protein is a major regulator of myeloid
homeostasis and neutrophil apoptosis.[19] A minority of patients with
HAX1-related neutropenia might have also neurodevelopmental delay and
epilepsy.[20] Biallelic (homozygous or compound heterozygous) G6PC3
pathogenic variants can cause a phenotypic spectrum that ranges from
nonsyndromic isolated severe congenital neutropenia to "classic"
neutropenia associated with cardiovascular and/or urogenital
abnormalities, endocrine dysfunctions, intermittent thrombocytopenia,
lymphopenia, thymic hypoplasia, recurrent bacterial infections, failure
to thrive and poor postnatal growth, which define the
glucose-6-phosphatase catalytic subunit 3 (G6PC3) deficiency, also
named Dursun syndrome.[21]
An X-linked form of congenital
neutropenia is caused by gain-of-function mutations in the WAS gene,
coding for the actin regulator WASp (Wiskott-Aldrich syndrome protein),
which are different from those causing Wiskott-Aldrich
thrombocytopenia, and defined by impaired cytoskeleton activity leading
to aberrant generation of neutrophils with reduced chemotactic
capacity.[22] Moreover, the Gfi-1 zinc finger transcriptional repressor
oncoprotein Gfi-1 has been related to myelopoiesis, and heterozygous
germline mutations in the GFI1 gene cause a severe form of congenital
neutropenia.[23] A different ethnic form of neutropenia linked to the
genetic deletion of the Duffy antigen receptor for chemokines
(DARC-null genotype) has been reported among Africans, having potential
effects on the development of different infectious diseases in these
individuals.[24] Additional diseases characterized by reduced white
blood cell count which do not have a genetic basis are benign familial
neutropenia, idiopathic neutropenia of unknown cause and autoimmune
neutropenia, for which anti-neutrophil antibodies need to be
demonstrated.
The Common Outlet of Infections in Congenital Neutropenia
ELANE-related
neutropenia represents a disease spectrum encompassing congenital
neutropenia, cyclic neutropenia, and intermediate findings between the
two phenotypes. Patients with cyclic neutropenia display a clinical
syndrome with fever, oral and mucosal ulcers or opportunistic
infections during the neutropenic phase. Stomatological infections are
very frequent after 2 years of age, and if neutropenia is severe they
are characterized by erosive, hemorrhagic or painful gingivitis
associated with aphthae and oral furuncles of the tongue and cheek
mucosa. Chronic and severe infections in the lung, liver or soft
tissues occurring at irregular intervals are more typical of severe
congenital neutropenia. Fever, malaise, oral aphthosis and mild sore
throat every three weeks is the usual presentation of cyclic
neutropenia in children: the typical onset is in the first year of
life. Symptoms may range from mild to severe, depending on the degree
and duration of neutropenia.[25] Infections of the
paranasal sinuses, upper- and lower respiratory tract and skin,
including the perianal area, may occur if the absolute neutrophil count
drops near to 0, and such an extremely low count may last for up to
3-to-5 days, giving rise to severe infections. Cellulitis may occur
during periods of neutropenia, even perianal cellulitis, but bacteremia
is infrequently proved. Abdominal pain and signs of acute abdomen,
suggesting sepsis and bacteremia from colonic ulcers, have been also
reported.
Tonsillitis, pharyngitis, gingivitis, swollen lymph
nodes, and dermatological infections are frequently encountered in
toddlers and older children. More than 60% of patients have skin and
pharyngeal symptoms, cervical lymphadenopathy, fever, and fatigue more
than 5 times a year. Some children might exhibit periodontitis with
alveolar bone fragility, and some may even display early loss of
permanent teeth.[26] Between the neutropenic periods,
children are generally healthy and grow well. Although congenital
neutropenia is usually discovered in childhood, the disease lasts
throughout the lifetime. However, the overall course of cyclic
neutropenia is benign, compared with other neutropenias.[27]
The systemic symptoms usually diminish after adolescence, but adult
patients may continue to experience oral ulcers, gingivitis,
periodontitis, and other mild infections.[28] Most infections are caused by common organisms lining on patients' body surfaces, including Clostridia
species and anaerobes of the intestinal microbiota. In contrast,
bacterial infections can be severe in congenital neutropenia and may
even occur in the neonatal period, when early-onset omphalitis could be
the first disease symptom. In addition to the risk of bacterial
infections, human papillomavirus infections can occur. The cumulative
incidence of hematologic malignancies is scarce in cyclic neutropenia,
differently from severe congenital neutropenia.[29]
The risk of developing myelodysplasia or acute myelogenous leukemia might vary considerably depending on the specific ELANE variant, and a consultation with a clinical geneticist should be warranted.[30]
In addition to leukemic transformation, solid tumors may also develop
early in life, such as kidney tumors and papilloma virus-induced
carcinoma. In the past, the risk of mortality for patients with
congenital neutropenia was related to the occurrence of necrotizing
enterocolitis, peritonitis, or sepsis involving Escherichia coli or Clostridium species.[31]
Today,
the development of blood malignancy is the major cause of mortality in
patients with congenital neutropenia, though the spectrum of somatic
mutations contributing to leukemic transformation has not been
characterized.[32] Patients who present with poor
growth and fatty stools need testing for the pancreatic function to
rule out Shwachman-Diamond syndrome, an autosomal recessive disorder
with multisystemic abnormalities, including exocrine pancreatic
insufficiency, short stature, and neutropenia (with hyposegmented
neutrophils); assessment of pancreatic function can be useful for
diagnosis.[33] A congenital form of neutropenia can
be also found in glycogen storage disease type 1b, in a specific form
of immunodeficiency with oculo-cutaneous albinism called
Chediak-Higashi syndrome (in which neutrophils contain abnormal
cytoplasmic granulations), in Griscelli syndrome type 2 (with partial
albinism) and in other metabolic diseases or bone marrow failure
syndromes. Table 1 lists the most relevant causes of congenital neutropenia and other conditions accompanied by neutropenia.
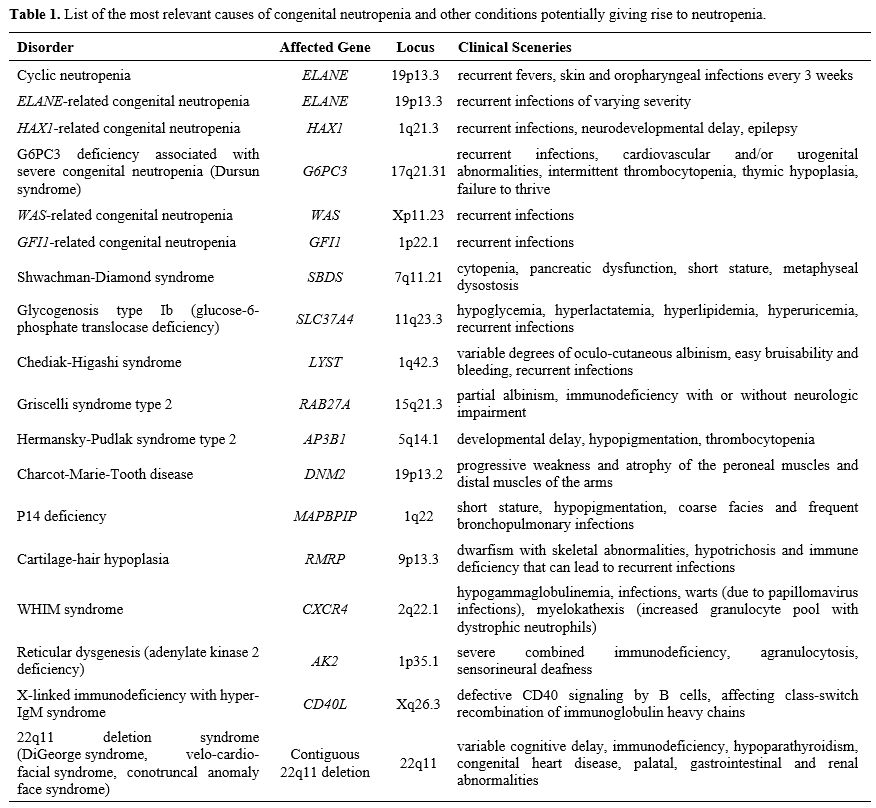 |
Table 1. List of the
most relevant causes of congenital neutropenia and other conditions
potentially giving rise to neutropenia. |
List of the most relevant causes of congenital neutropenia and other conditions potentially giving rise to neutropenia.
Post-infectious
and drug-related are the most frequent etiologies of neutropenia, but
they have no frankly specific features. The congenital neutropenia
syndromes are taken into consideration if there are recurrent
infections in patient's history, if neutropenia is severe, and if any
associated anomalies suggest a genetic disorder. Diagnosis of cyclic
neutropenia depends on serial measurements of the absolute neutrophil
count over several weeks: all affected children have a neutrophil count
below 200/mm3 for three-to-five days
at approximately three-week intervals. This disease is usually
diagnosed within the first year of life, based on the pattern of
recurrent fevers, skin inflammation, and oral ulcerations with serial
blood cell count assessment. Bone marrow examination is not needed for
establishing the diagnosis, but should be performed to rule out
malignant hemopathies in the case of additional hematological
abnormalities; oscillations of other cells such as lymphocytes,
eosinophils, and platelets may also be observed. A reciprocal increase
in blood monocytes and reticulocytes can occur during the neutrophil
nadir.[34]
A proband with a suggestive clinical
scenery that recurs over time requires genetic testing, and diagnosis
follows the identification of one heterozygous pathogenic variant in
the ELANE gene. Testing a
panel of genes rather than a single gene may be useful since distinct
genetic disorders can be associated with variably cycling neutrophil
counts. The differential diagnosis between severe congenital
neutropenia and cyclic neutropenia is important, as the severity of
infections may be higher in the first and because of the risk of
developing potentially life-threatening diseases, such as
myelodysplastic syndrome and acute myeloid leukemia.[35,36] Cyclic neutropenia is usually not associated with malignant transformation to hematologic cancer, except for very few cases.[37,38]
Rosenberg et al. studied the risk of sepsis mortality and incidence of
leukemia in a population of 374 people with severe congenital
neutropenia, receiving long-term therapy with G-CSF, finding that
mortality due to sepsis was stable at 0.9% per year, while the risk of
developing myelodysplastic syndrome or acute myeloid leukemia increased
significantly over time, from 2.9% per year after 6 years to 8% per
year after 12 years of therapy with G-CSF.[39,40]
Investigations such as complete blood cell count, bone marrow biopsy,
tumoral markers, serum level of G-CSF, chest X-ray or chest ultrasound,
and computerized tomography scan of the chest should be done to rule
out other immunodeficiency disorders. Autoinflammatory disorders should
be also considered if the recurrent episodes of fever are combined with
organ-specific sterile inflammatory manifestations.[41]
In children aged 1-3 years with neutropenia not-caused by cyclic
neutropenia or Kostmann syndrome, the presence of neutrophil-specific
autoantibodies results in a peripheral destruction of neutrophils.
Although these infants lack peripheral blood neutrophils, they usually
do not suffer from severe infections.[42] Table 2 shows some critical hints for diagnosing congenital neutropenia.
 |
Table 2. Pivotal keys to the diagnosis of congenital neutropenia in children. |
Assessing the Risk of Evolution to Hematologic Malignancy
Myelodysplastic
syndromes are diseases affecting patients prevalently over 65 years. In
contrast, the genetic susceptibility to myelodysplastic syndrome and
acute myeloid leukemia can be rarely demonstrated in children. A
malignant progression of congenital neutropenia has seldom been seen in
the pre-growth factor era, but the number of patients progressing to
blood cancer has increased with the improved life expectancy achieved
after G-CSF introduction as treatment. The development of
myelodysplastic syndrome and acute myeloid leukemia in congenital
neutropenia remains the major cause of mortality in these patients.[43] This risk is shared by both ELANE-related and HAX1-related severe congenital neutropenia, and specific ELANE mutations (e.g. G214R and C151Y) have a higher risk of progression to acute myeloid leukemia.[44]
Apart from allogeneic hematopoietic stem cell transplantation,
treatment options are limited, and there are neither reliable
biomarkers that predict progression, nor effective prevention
strategies.
Myelodysplastic syndromes secondary to severe
congenital neutropenia are frequently associated with monosomy 7 and
abnormalities of chromosome 21, which are relatively uncommon in de novo
myeloid leukemia. The accumulation of mutations in hematopoietic stem
cells with increasing age results in the production of a genetically
heterogeneous cell population, with each stem cell possessing its own
unique set of private mutations. Selective clonal hematopoiesis due to
mutations in the tumor suppressor TP53
gene has been found in almost 50% of patients with Shwachman-Diamond
syndrome, but not in those with severe congenital neutropenia: in
particular, Shwachman-Diamond patients have an impaired ribosome
biogenesis driving the expansion of hematopoietic stem cells which
carry TP53 mutations. Factors
that increase the rate at which mutations accumulate in stem cells may
increase the frequency of clonal hematopoiesis and blood cancer
uprising. The acquisition of TP53
mutations can be framed as an early event for the transformation of
Shwachman-Diamond-related neutropenia into myelodysplastic syndrome or
acute myeloid leukemia.[45]
Different neutropenic patients with risk of leukemic progres¬sion can show hematopoietic clones with somatic mutations in the CSF3R
gene, encoding the G-CSF receptor, resulting in a truncated form of the
G-CSF receptor, leading to defective internalization and aberrant
signaling properties of the receptor: these clones may persist for
months and even years before myelodysplastic syndrome or acute myeloid
leukemia becomes overt. Of note, no increase in clonal hematopoiesis
due to other gene mutations can be observed, demonstrating the highly
selective nature of CSF3R-dependent clonal expansion in severe congenital neutropenia and clearly suggesting that CSF3R mutations contribute to the development of myelodysplastic syndrome and acute myeloid leukemia.[46]
In
recent times, mutations in some oncogenes have been linked to familial
myelodysplastic syndrome and acute myeloid leukemia. For instance, the GATA2
gene, located at the chromosome 3, encoding for a nuclear transcription
factor, has been identified as a potential trigger to develop blood
malignancy in neutropenic patients, mostly if germline GATA2 mutations are associated with partial or complete deletion of chromosome 7.[47] Somatic RUNX1 mutations on chromosome 21 have been found in approximately 10% of patients with de novo
acute myeloid leukemia, but are more common in secondary forms of
myelodysplastic syndrome and acute myeloid leukemia, mostly if
originating from certain types of leukemia-prone neutropenic syndromes.
How RUNX1 mutations and how
the Runt-related transcription factor contribute to the pathobiology of
secondary hemopathies, often characterized by adverse prognosis and
re¬fractoriness to treatment, is still unknown.[48]
Therefore,
although we have gained significant knowledge of the heterogeneous
genetic origin of congenital neutropenia, it is still undeciphered how
these mutations predispose to leukemia and molecular mechanisms
eliciting the transformation of neutropenia to myelodysplastic syndrome
or acute myeloid leukemia are poorly understood. Additional treatment
strategies using small molecule inhibitors with selectivity for the
mutant genes to eradicate the mutant clones shortly after they appear
or gene therapies correcting the underlying genetic defect in
hematopoietic stem cells should be explored in real-life sceneries.
How to Put Children with Recurrent Fevers in Differential Diagnosis
Cyclic
neutropenia represents a diagnostic dilemma, as most patients exhibit
non-specific signs or symptoms dominated by recurrent fevers.
Evaluation of children with cyclic neutropenia is based on the
recognition of pivotal recurrent symptoms, disease duration, history of
hereditary inheritance, and periodic assessment of the leukocyte count
to prompt a differential diagnosis.[49,50] This
clinical picture should remind several causes of recurring fevers in
pediatrics, such as recurrent tonsillitis, infectious diseases, and
Behçet's disease, but diagnosis of cyclic neutropenia is confirmed if
the periodic oscillation of neutrophil count (every 21 days) is
demonstrated at a sufficient number of time points (at least three
times per week over 6 weeks), which is sometimes challenging for
children.[51] Many diseases share recurrent fever as
a common presenting feature in childhood: in particular, if fevers are
not truly periodic (i.e., do not have a quite regular interval between
febrile episodes) the monogenic periodic fever syndromes, caused by
activation of the innate immune system, should be scrutinized.[52]
These syndromes are different expressions of a primary dysfunction of
innate immunity and are collectively named "autoinflammatory
disorders", complex and heterogeneous diseases in which there is no
evidence of adaptive immunity involvement, neither high-titre
autoantibodies nor antigen-specific T cells, and no relationship with
infectious triggers.[53] Children with hereditary
autoinflammatory disorders display periodically-recurring features
consisting of fever and organ-specific inflammation, with symptom-free
intervals of different duration between attacks. Among
autoinflammatory disorders, familial Mediterranean fever and mevalonate
kinase deficiency need to be taken into consideration as their
manifestations may overlap with those of cyclic neutropenia.[54]
The first is characterized by periodic short-lasting febrile episodes
combined with serositis and/or arthritis in patients of Arabian,
Armenian, non-Ashkenazi Jewish or Turkish descent.[55,56]
The second is characterized by variably recurrent self-limiting febrile
episodes of 3-7 days combined with concurrent debilitating symptoms
which involve the muco-cutaneous, gastrointestinal and musculoskeletal
system and last about one week.[57,58] On the other
hand, recurrent flares of longlasting fever combined with recurrent
inflammation in the muscles, joints, gastrointestinal tube and skin are
the leading features of tumor necrosis factor receptor-associated
periodic syndrome.[59] There is no specific
laboratory examination to support the diagnosis of autoinflammatory
disorders, except for the genetic analysis; their onset is usually
during the first decade in about 50% of cases and symptoms may also
start in the first months of life, requiring a strict differentiation
with other childhood emergencies.[60] During
a typical attack of autoinflammatory disorders blood tests show a
generalized increase of inflammatory parameters with a parallel
neutrophil leukocytosis (until and over 20.000/mm3).[61]
This feature should help clinicians in differentiating autoinflammatory
disorders from neutropenia, in which cyclic and "sterile" inflammatory
phenomena outside the oral cavity are not usually encountered. Chronic
inflammation in autoinflammatory disorders may also cause irreversible
damage in multiple organ systems, such as visual loss, deafness, joint
restriction and amyloidosis.[62] Conversely,
the periodic fever, aphthous stomatitis, pharyngitis and cervical
adenitis (or PFAPA) syndrome, the most common non-inherited cause of
periodic fever in childhood, is very similar to cyclic neutropenia and
similarly may affect patient's family quality of life.[63]
PFAPA children display inflammatory symptoms restricted to the
oropharyngeal and neck lymphoid tissue with "clockwork" regularity
every to 3-to-6 weeks: their fevers lasting 3-to-6 days recur combined
with aphthous ulcers, pharyngitis and/or tonsillitis and cervical lymph
node enlargement.[64] In contrast with cyclic
neutropenia, these children have high white blood cell counts with
preponderance of neutrophils and high levels of inflammatory markers
during febrile episodes, while the neutrophil count turns to normal in
the interfebrile periods.[65] Procalcitonin, a
significant marker of bacterial infections, does not increase during
PFAPA attacks, while serum immunoglobulins are usually normal during
attacks.[66] The discrimination between PFAPA
syndrome and cyclic neutropenia may be particularly challenging, as it
requires a painstaking collection of clinical and laboratory data, and
this challenge also involves the area of internal medicine, as PFAPA
symptoms have been reported even in adulthood.[67-69]
There is a need for universal diagnostic criteria of PFAPA syndrome,
which can be valid for both children and adults. Febrile flares of
mevalonate kinase deficiency can closely resemble PFAPA syndrome and
cyclic neutropenia, but the presence of significant diarrhea or
vomiting, lymphadenopathy outside of the cervical area and episodes
triggered by immunizations may steer the diagnosis towards mevalonate
kinase deficiency.[70] Table 3
lists the most relevant general features of hereditary periodic fever
syndromes and PFAPA syndrome, which require consideration in the
differential diagnosis with cyclic neutropenia. Unfortunately, many
questions related to PFAPA pathogenesis or a potential genetic basis
for PFAPA symptoms and the reason why inflammation is localized at the
oral cavity and neck, similar to cyclic neutropenia, remain unsolved.
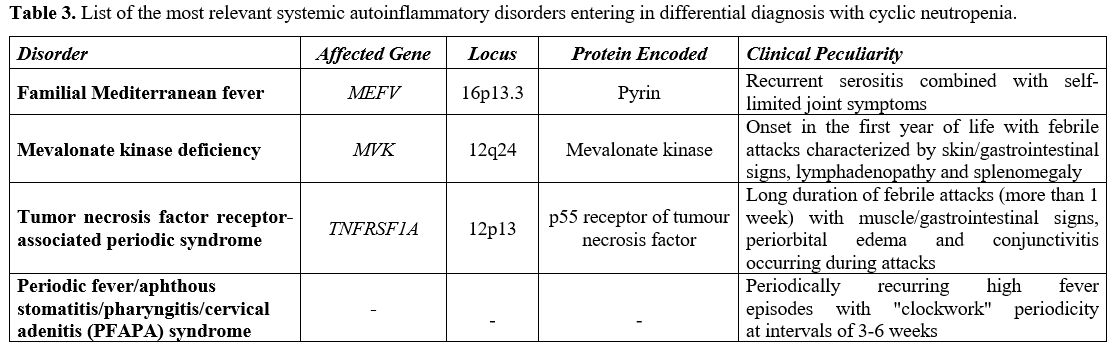 |
Table 3. List of the most
relevant systemic autoinflammatory disorders entering in differential
diagnosis with cyclic neutropenia. |
Clues for the Specific Management of Children with Congenital Neutropenia
Over
the past years, different hematologists have collaborated to optimize
the management of patients with congenital neutropenia, finding that a
multidisciplinary approach including immunologists, radiologists, and
dentists is crucial through regular patients’ follow-up visits to
improve the overall outcome. Treatment of severe chronic neutropenia
should focus on the prevention of infections, managing organ
dysfunction, and preventing leukemic transformation.[71]
All fevers and infections require prompt evaluation and treatment,
while abdominal pain requires excluding peritonitis. Severe infections
are rare in cyclic neutropenia, and most infections usually respond to
antibiotics. Longer-term follow-up studies of patients with cyclic
neutropenia showed that the most relevant complications were
spontaneous peritonitis, segmental bowel necrosis, and septicemia,
though they occurred rarely.[72] Children
suffering from severe congenital neutropenia require long-term
administration of recombinant human G-CSF, given by subcutaneous
injections daily or every other day, which is also the same treatment
for patients suffering from cyclic neutropenia. Treatment with G-CSF
(either filgrastim or pegfilgrastim) is safe and well-tolerated and is
effective for elevating the neutrophil count in both congenital and
cyclic neutropenia, ameliorating disease symptoms and reducing most
infectious complications.[73] In cyclic neutropenia
G-CSF shortens the periods of neutropenia as well as the length of the
neutropenic cycle: once the absolute neutrophil count normalizes, the
resistance to infections improves. Common side effects of G-CSF include
bone pain, headache, splenomegaly, and osteoporosis, but no adverse
effects on growth and development have been reported in children.
Therefore, G-CSF treatment enables patient's participation to school
and recreational activities without any concerns. The optimal dosage of
filgrastim in cyclic neutropenia is 2-to-3 μg/kg/day: this dose is
lower than that used for severe congenital neutropenia (5-10 μg/kg/day) or for chemotherapy-associated neutropenia, and may require adjustments based on the therapeutic response.[74]
Treatment combined with broad-spectrum antibiotics is important and
even lifesaving in the case of complicated serious infections, which
may be caused by mixed anaerobic/aerobic pathogens. Trying to
synchronize filgrastim treatment to coincide with the period of
neutropenia in cyclic neutropenia is hard, but administrations of G-CSF
will usually change the periodicity of neutropenia. The drug is
effective from as early as 6 months of age and treatment should
continue lifelong, as needed. The main goal of treatment is to maintain
the nadir of the absolute neutrophil count over 500/mm3,
which substantially reduces the risk of infections and affects health
of the oral cavity, improving both masticatory ability and comfortable
eating.[75] The long-acting pegylated formulation of
G-CSF, pegfilgrastim, is also effective and convenient, but is
difficult to dose in children and often leads to more severe bone pain
as a side effect.[76] Although granulocyte-macrophage
colony-stimulating factor has been administered for severe chronic
neutropenia, it is less effective than G-CSF and associated with more
adverse effects.[77] It has been found that in JAGN1-mutant
granulocytes, characterized by ultrastructural defects, paucity of
granules, aberrant glycosylation of multiple proteins and increased
apoptosis, in which the production of the JAGN1 protein located in the
endoplasmic reticulum is deficient, the response to G-CSF may be
poorer.[78] As
children grow and become adults, the dose of G-CSF should be adjusted
in accordance with symptoms and blood cell count, rather than depending
on the body weight. Because levels of neutrophils fluctuate in cyclic
neutropenia, the neutrophil count has to be periodically monitored for
several weeks after starting G-CSF. Sometimes, neutrophil cycling may
be replaced by a mild chronic neutropenia in the third decade. An old
report suggested that a combination therapy of G-CSF and high-dose
immunoglobulins could be effective to induce the disappearance of
neutrophil oscillations in cyclic neutropenia, but this therapeutic
protocol has not been confirmed.[79] Lastly,
hematopoietic stem cell transplantation is the ultimate radical
treatment for congenital neutropenia, as it can permanently correct the
disease, being the long-term option for patients who do not respond to
G-CSF.[80] Because G-CSF may promote leukemic
transformation, patients requiring higher doses of G-CSF to prevent
infections are candidates for stem cell transplantation, while patients
not-undergoing transplantation require long-term surveillance to reveal
a malignant transformation of the disease. Reassuringly, there are
preclinical data giving proof of concept for treatment of congenital
neutropenia through autologous transplantation of gene-edited cells for
patients who did not show any response to G-CSF and for whom bone
marrow transplantation had failed.[81] Supportive
care is also important to reduce the risk and severity of infections
and should include oral hygiene and dental care through regular health
assessments at intervals of 6 months and twice-daily oral rinsing with
chlorhexidine after tooth brushing.[82] Bone density
should be checked for the risk of osteoporosis during prolonged
treatment with G-CSF, as osteoporosis is a possible side effect of this
medication, and monitoring vitamin D levels should be also provided.[83]
Furthermore, it is important to maintain an age-appropriate schedule of
immunizations, while there is no need to avoid public places with
people aggregation since most infections are caused by resident
organisms from the intestinal microbiota. Common viral infections may
be complicated with bacterial infections; however, prophylactic
antibiotics are not recommended, as this might eventually select
resistant organisms.[84]
General Conclusive Remarks
Despite
the great steps in understanding congenital neutropenia, the
constellation of clinical symptoms and recurrent fevers occurring in
this protean disease generate frustration and grievous interactions
among the clinician and patient's family members. In conclusion, among
the rare primary disorders of the hematopoietic stem cell, it is
crucial to consider cyclic neutropenia, transmitted with autosomal
dominant inheritance, which is characterized by a biological clock
responsible for sinusoidal variations of the neutrophil count every 21
days and recurrent infections. This disorder is distinct from permanent
neutropenia, the classic feature of Kostmann syndrome, which starts in
infancy. Infectious complications of cyclic neutropenia are rarer than
those occurring in the severe congenital neutropenia. However,
treatment with G-CSF can be largely effective, though the dose required
to normalize neutrophils may vary. A strict differentiation of cyclic
neutropenia from hereditary monogenic autoinflammatory disorders and
PFAPA syndrome is mandatory in children.
References
- Badolato R, Fontana S, Notarangelo LD, et al.
Congenital neutropenia: advances in diagnosis and treatment. Curr Opin
Allergy Clin Immunol 2004;4:513-21. https://doi.org/10.1097/00130832-200412000-00007 PMid:15640692
- Segel GB, Halterman JS. Neutropenia in pediatric practice. Pediatr Rev 2008;29:12-23. https://doi.org/10.1542/pir.29-1-12 PMid:18166617
- Dale
DC, Person RE, Bolyard AA, et al. Mutations in the gene encoding
neutrophil elastase in congenital and cyclic neutropenia. Blood
2000;96:2317-22. https://doi.org/10.1182/blood.V96.7.2317 PMid:11001877
- Dale DC, Hammond WP. Cyclic neutropenia: a clinical review. Blood Rev 1988;2:178-85. https://doi.org/10.1016/0268-960X(88)90023-9 PMid: 3052663
- Horwitz
M, Benson KF, Person RE, et al. Mutations in ELA2, encoding neutrophil
elastase, define a 21-day biological clock in cyclic haematopoiesis.
Nat Genet 1999;23:433-6. https://doi.org/10.1038/70544 PMid:10581030
- Rigante D. When, how, and why do fevers hold children hostage? J Evid Based Med 2020;13:85-8. https://doi.org/10.1111/jebm.12377 PMid:32086995
- Kollner
I, Sodeik B, Schreek S, et al. Mutations in neutrophil elastase causing
congenital neutropenia lead to cytoplasmic protein accumulation and
induction of the unfolded protein response. Blood 2006;108:493-500. https://doi.org/10.1182/blood-2005-11-4689 PMid:16551967
- Ancliff PJ, Gale RE, Linch DC. Neutrophil elastase mutations in congenital neutropenia. Hematology 2003;8:165-71. https://doi.org/10.1080/1024533031000107497 PMid:12745650
- Garwicz
D, Lennartsson A, Jacobsen SE, et al. Biosynthetic profiles of
neutrophil serine proteases in a human bone marrow-derived cellular
myeloid differentiation model. Haematologica 2005;90:38-44. PMid:
15642667
- Haurie C, Dale DC, Mackey MC.
Cyclical neutropenia and other periodic hematological disorders: a
review of mechanisms and mathematical models. Blood 1998;92:2629-40. https://doi.org/10.1182/blood.V92.8.2629.420a35_2629_2640 PMid:9763544
- Mir
P, Klimiankou M, Findik B, et al. New insights into the pathomechanism
of cyclic neutropenia. Ann NY Acad Sci 2020;1466:83-92. https://doi.org/10.1111/nyas.14309 PMid:32083314
- Makaryan
V, Zeidler C, Bolyard AA, et al. The diversity of mutations and
clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol
2015;22:3-11. https://doi.org/10.1097/MOH.0000000000000105 PMid:25427142 PMCid:PMC4380169
- Olofsen
PA, Bosch DA, Roovers O, et al. PML-controlled responses in severe
congenital neutropenia with ELANE-misfolding mutations. Blood Adv
2021;5:775-86. https://doi.org/10.1182/bloodadvances.2020003214 PMid:33560392 PMCid:PMC7876869
- Boxer LA, Newburger PE. A molecular classification of congenital neutropenia syndromes. Pediatr Blood Cancer 2007;49:609-14. https://doi.org/10.1002/pbc.21282 PMid:17584878
- Ishikawa
N, Okada S, Miki M, et al. Neurodevelopmental abnormalities associated
with severe congenital neutropenia due to the R86X mutation in the HAX1
gene. J Med Genet 2008;45:802-7. https://doi.org/10.1136/jmg.2008.058297 PMid:18611981
- Aprikyan
AAG, Kutyavin T, Stein S, et al. Cellular and molecular abnormalities
in severe congenital neutropenia predisposing to leukemia. Exp Hematol
2003;31:372-81. https://doi.org/10.1016/S0301-472X(03)00048-1
- Newburger
PE, Pindyck TN, Zhu Z, et al. Cyclic neutropenia and severe congenital
neutropenia in patients with a shared ELANE mutation and paternal
haplotype: evidence for phenotype determination by modifying genes.
Pediatr Blood Cancer 2010;55:314-17. https://doi.org/10.1002/pbc.22537 PMid:20582973 PMCid:PMC2913300
- Horwitz
MS, Corey SJ, Grimes HL, et al. ELANE mutations in cyclic and severe
congenital neutropenia: genetics and pathophysiology. Hematol Oncol
Clin North Am 2013;27:19-41. https://doi.org/10.1016/j.hoc.2012.10.004 PMid:23351986 PMCid:PMC3559001
- Klein
C, Grudzien M, Appaswamy G, et al. HAX1 deficiency causes autosomal
recessive severe congenital neutropenia (Kostmann disease). Nature
Genet 2007;39:86-92.https://doi.org/10.1038/ng1940 PMid:17187068
- Bellanné-Chantelot
C, Clauin S, Leblanc T, et al. Mutations in the ELA2 gene correlate
with more severe expression of neutropenia: a study of 81 patients from
the French Neutropenia Register. Blood 2004;103:4119-25. https://doi.org/10.1182/blood-2003-10-3518 PMid:14962902
- Banka S, Newman WG, Özgül RK, et al. Mutations in the G6PC3 gene cause Dursun syndrome. Am J Med Genet A 2010;152A:2609-11. https://doi.org/10.1002/ajmg.a.33615 PMid:20799326
- Devriendt
K, Kim AS, Mathijs G, et al. Constitutively activating mutation in WASP
causes X-linked severe congenital neutropenia. Nat Genet 2001;27:313-7.
https://doi.org/10.1038/85886 PMid:11242115
- Person
RE, Li FQ, Duan Z, et al. Mutations in proto-oncogene GFI1 cause human
neutropenia and target ELA2. Nat Genet 2003;34:308-12. https://doi.org/10.1038/ng1170 PMid:12778173 PMCid:PMC2832179
- Thobakgale CF, Ndung'u T. Neutrophil counts in persons of African origin. Curr Opin Hematol 2014;21:50-7. https://doi.org/10.1097/MOH.0000000000000007 PMid:24257098
- Guerry D, Dale DC, Omine M, et al. Periodic hematopoiesis in human cyclic neutropenia. J Clin Invest 1973;52:3220-30. https://doi.org/10.1172/JCI107522 PMid:4750451 PMCid:PMC302598
- Germeshausen
M, Deerberg S, Peter Y, et al. The spectrum of ELANE mutations and
their implications in severe congenital and cyclic neutropenia. Hum
Mutat 2013;34:905-14. https://doi.org/10.1002/humu.22308 PMid:23463630
- Wright
DG, Dale DC, Fauci AS, et al. Human cyclic neutropenia: clinical review
and long-term follow-up of patients. Medicine (Baltimore) 1981;60:1-13.
https://doi.org/10.1097/00005792-198101000-00001 PMid:7453561
- Palmer
SE, Stephens K, Dale DC. Genetics, phenotype, and natural history of
autosomal dominant cyclic hematopoiesis. Am J Med Genet 1996;66:413-22.
https://doi.org/10.1002/(SICI)1096-8628(19961230)66:4<413::AID-AJMG5>3.0.CO;2-L
- Rosenberg
PS, Alter BP, Link DC, et al. Neutrophil elastase mutations and risk of
leukaemia in severe congenital neutropenia. Br J Haematol
2008;140:210-3. https://doi.org/10.1111/j.1365-2141.2007.06897.x PMid:18028488 PMCid:PMC3143022
- Shu
Z, Li XH, Bai XM, et al. Clinical characteristics of severe congenital
neutropenia caused by novel ELANE gene mutations. Pediatr Infect Dis J
2015;34:203-7. https://doi.org/10.1097/INF.0000000000000522 PMid:25162927
- Barnes
C, Gerstle JT, Freedman MH, et al. Clostridium septicum myonecrosis in
congenital neutropenia. Pediatrics 2004;114:e757-60. https://doi.org/10.1002/(SICI)1096-8628(19961230)66:4<413::AID-AJMG5>3.0.CO;2-L PMid: 8989458
- Link DC. Mechanisms of leukemic transformation in congenital neutropenia. Curr Opin Hematol 2019;26:34-40. https://doi.org/10.1097/MOH.0000000000000479 PMid:30431463 PMCid:PMC6447304
- Mercuri
A, Cannata E, Perbellini O, et al. Immunophenotypic analysis of
hematopoiesis in patients suffering from Shwachman-Bodian-Diamond
syndrome. Eur J Haematol 2015;95:308-15. https://doi.org/10.1111/ejh.12490
- PMid:25402872Dale DC, Welte K. Cyclic and chronic neutropenia. Cancer Treat Res 2011;157:97-108. https://doi.org/10.1007/978-1-4419-7073-2_6 PMid:21052952
- Rosenberg
PS, Zeidler C, Bolyard AA, et al. Stable long-term risk of leukaemia in
patients with severe congenital neutropenia maintained on G-CSF
therapy. Br J Haematol 2010;150:196-9. https://doi.org/10.1111/j.1365-2141.2010.08216.x PMid:20456363 PMCid:PMC2906693
- Donadieu
J, Leblanc T, Bader Meunier B, et al. Analysis of risk factors for
myelodysplasias, leukemias and death from infection among patients with
congenital neutropenia. Experience of the French Severe Chronic
Neutropenia Study Group. Haematologica 2005;90:45-53. PMid: 15642668
- Zeidler C, Welte K. Kostmann syndrome and severe congenital neutropenia. Semin Hematol 2002;39:82-8. https://doi.org/10.1053/shem.2002.31913 PMid:11957189
- Klimiankou
M, Mellor-Heineke S, Klimenkova O, et al. Two cases of cyclic
neutropenia with acquired CSF3R mutations, with 1 developing AML. Blood
2016;127:2638-41. https://doi.org/10.1182/blood-2015-12-685784 PMid:27030388
- Rosenberg
PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and
mortality from sepsis in patients with severe congenital neutropenia
receiving long-term G-CSF therapy. Blood 2006; 107:4628-35. https://doi.org/10.1182/blood-2005-11-4370 PMid:16497969 PMCid:PMC1895804
- Dale DC, Bolyard A, Marrero T, et al. Long-term effects of G-CSF therapy in cyclic neutropenia. N Engl J Med 2017;377:2290-2. https://doi.org/10.1056/NEJMc1709258 PMid:29211670 PMCid:PMC5777346
- De
Rose DU, Coppola M, Gallini F, Maggi L, Vento G, Rigante D. Overview of
the rarest causes of fever in newborns: handy hints for the
neonatologist. J Perinatol 2021;41:372-82. https://doi.org/10.1038/s41372-020-0744-8 PMid:32719496
- Bux
J, Behrens G, Jaeger G, et al. Diagnosis and clinical course of
autoimmune neutropenia in infancy: analysis of 240 cases. Blood
1998;91:181-6. https://doi.org/10.1182/blood.V91.1.181.181_181_186 PMid:9414283
- Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia-a review. Br J Haematol 2008;140:123-32. https://doi.org/10.1111/j.1365-2141.2007.06909.x PMid:18173751
- Makaryan
V, Zeidler C, Bolyard AA, et al. The diversity of mutations and
clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol
2015;22:3-11. https://doi.org/10.1097/MOH.0000000000000105 PMid:25427142 PMCid:PMC4380169
- Xia J, Miller CA, Baty J, et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood 2018;131:408-16. https://doi.org/10.1182/blood-2017-08-801985 PMid:29092827 PMCid:PMC5790127
- Freedman
MH, Bonilla MA, Fier C, et al. Myelodysplasia syndrome and acute
myeloid leukemia in patients with congenital neutropenia receiving
G-CSF therapy. Blood 2000;96:429-36. PMid: 10887102
- Pasquet
M, Bellanné-Chantelot C, Tavitian S, et al. High frequency of GATA2
mutations in patients with mild chronic neutropenia evolving to MonoMac
syndrome, myelodysplasia, and acute myeloid leukemia. Blood
2013;121:822-9. https://doi.org/10.1182/blood-2012-08-447367 PMid:23223431 PMCid:PMC3714670
- Olofsen PA, Touw IP. RUNX1 mutations in the leukemic progression of severe congenital neutropenia. Mol Cells 2020;43:139-44. https://doi.org/10.14348/molcells.2020.0010 PMid: 32041395
- Rigante
D. Autoinflammatory syndromes behind the scenes of recurrent fevers in
children. Med Sci Monit 2009;15:RA179-87. PMid: 19644432
- Donadieu
J, Beaupain B, Fenneteau O, et al. Congenital neutropenia in the era of
genomics: classification, diagnosis, and natural history. Br J Haematol
2017;179:557-74. https://doi.org/10.1111/bjh.14887 PMid:28875503
- Marshall GS. Prolonged and recurrent fevers in children. J Infect 2014;68 Suppl 1:S83-93. https://doi.org/10.1016/j.jinf.2013.09.017 PMid:24120354
- Rigante
D. Phenotype variability of autoinflammatory disorders in the pediatric
patient: a pictorial overview. J Evid Based Med 2020;13:227-45. https://doi.org/10.1111/jebm.12406 PMid:32627322
- Rigante D. New mosaic tiles in childhood hereditary autoinflammatory disorders. Immunol Lett 2018;193:67-76. https://doi.org/10.1016/j.imlet.2017.11.013 PMid:29198619
- Rigante
D, Frediani B, Galeazzi M, et al. From the Mediterranean to the sea of
Japan: the transcontinental odyssey of autoinflammatory diseases.
Biomed Res Int 2013;2013:485103. https://doi.org/10.1155/2013/485103 PMid:23971037 PMCid:PMC3736491
- Manna
R, Rigante D. Familial Mediterranean fever: assessing the overall
clinical impact and formulating treatment plans. Mediterr J Hematol
Infect Dis 2019;11:e2019027. https://doi.org/10.4084/mjhid.2019.027 PMid:31205631 PMCid:PMC6548206
- Cantarini
L, Lopalco G, Selmi C, et al. Autoimmunity and autoinflammation as the
yin and yang of idiopathic recurrent acute pericarditis. Autoimmun Rev
2015;14:90-7. https://doi.org/10.1016/j.autrev.2014.10.005 PMid:25308531
- Esposito
S, Ascolese B, Senatore L, et al. Current advances in the understanding
and treatment of mevalonate kinase deficiency. Int J Immunopathol
Pharmacol 2014;27:491-8. https://doi.org/10.1177/039463201402700404 PMid:25572728
- Rigante
D. The protean visage of systemic autoinflammatory syndromes: a
challenge for inter-professional collaboration. Eur Rev Med Pharmacol
Sci 2010;14:1-18. PMid: 20184084
- Rigante
D, Lopalco G, Vitale A, et al. Key facts and hot spots on tumor
necrosis factor receptor-associated periodic syndrome. Clin Rheumatol
2014;33:1197-207. https://doi.org/10.1007/s10067-014-2722-z PMid:24935411
- Rigante
D. The broad-ranging panorama of systemic autoinflammatory disorders
with specific focus on acute painful symptoms and hematologic
manifestations in children. Mediterr J Hematol Infect https://doi.org/10.4084/mjhid.2018.067 PMid:30416699 PMCid:PMC6223578
- Rigante
D. A systematic approach to autoinflammatory syndromes: a spelling
booklet for the beginner. Expert Rev Clin Immunol 2017;13:571-97. https://doi.org/10.1080/1744666X.2017.1280396 PMid:28064547
- Ter
Haar NM, Annink KV, Al-Mayouf SM, et al. Development of the
autoinflammatory disease damage index (ADDI). Ann Rheum Dis
2017;76:821-30. https://doi.org/10.1136/annrheumdis-2016-210092 PMid:27811147
- Esposito
S, Bianchini S, Fattizzo M, et al. The enigma of periodic fever,
aphthous stomatitis, pharyngitis and adenitis syndrome. Pediatr Infect
Dis J 2014;33:650-2. https://doi.org/10.1097/INF.0000000000000346 PMid:24642520
- Rigante D. A developing portrait of hereditary periodic fevers in childhood. Expert Opin Orphan Drugs 2018;6:47-55. https://doi.org/10.1080/21678707.2018.1406797
- Manna R, Rigante D. The everchanging framework of autoinflammation. Intern Emerg Med 2021;16:1759-70. https://doi.org/10.1007/s11739-021-02751-7 PMid:33999387 PMCid:PMC8502124
- Yazgan
H, Keleş E, Yazgan Z, et al. C-reactive protein and procalcitonin
during febrile attacks in PFAPA syndrome. Int J Pediatr
Otorhinolaryngol 2012;76:1145-7. https://doi.org/10.1016/j.ijporl.2012.04.022 PMid:22658448
- Rigante
D, Corina L. Periodic fever, aphthous stomatitis, pharyngitis and
cervical adenitis (PFAPA) syndrome: a debate about diagnosis and
treatment in children continues. Int J Pediatr Otorhinolaryngol
2020;130:109830. https://doi.org/10.1016/j.ijporl.2019.109830 PMid:31866107
- Sicignano
LL, Rigante D, Moccaldi B, et al. Children and adults with PFAPA
syndrome: similarities and divergences in a real-life clinical setting.
Adv Ther 2021;38:1078-93. https://doi.org/10.1007/s12325-020-01576-8 PMid:33315168
- Rigante
D, Vitale A, Natale MF, et al. A comprehensive comparison between
pediatric and adult patients with periodic fever, aphthous stomatitis,
pharyngitis, and cervical adenopathy (PFAPA) syndrome. Clin Rheumatol
2017;36:463-8. https://doi.org/10.1007/s10067-016-3317-7 PMid:27251674
- Gentileschi
S, Vitale A, Frediani B, et al. Challenges and new horizons in the
periodic fever, aphthous stomatitis, pharingitis and adenitis (PFAPA)
syndrome. Expert Opin Orphan Drugs 2017;5:165-71. https://doi.org/10.1080/21678707.2017.1279049
- Pernu
HE, Pajari UH, Lanning M. The importance of regular dental treatment in
patients with cyclic neutropenia. Follow-up of 2 cases. J Periodontol
1996;67:454-9. https://doi.org/10.1902/jop.1996.67.4.454 PMid:8708974
- Geelhoed GW, Kane MA, Dale DC, et al. Colon ulceration and perforation in cyclic neutropenia. J Pediatr Surg 1973;8:379-82. https://doi.org/10.1016/0022-3468(73)90105-X
- Horwitz MS, Duan Z, Korkmaz B, et al. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood 2007;109:1817. https://doi.org/10.1182/blood-2006-08-019166 PMid:17053055 PMCid:PMC1801070
- Dale
DC, Cottle TE, Fier CJ, et al. Severe chronic neutropenia: treatment
and follow-up of patients in the Severe Chronic Neutropenia
International Registry. Am J Hematol 2003;72:82-93. https://doi.org/10.1002/ajh.10255 PMid:12555210
- Hammond
WP, Price TH, Souza LM, et al. Treatment of cyclic neutropenia with
granulocyte colony-stimulating factor. N Engl J Med 1989;320:1306-11. https://doi.org/10.1056/NEJM198905183202003 PMid:2469956
- Matarasso
S, Daniele V, Iorio Siciliano V, et al. The effect of recombinant
granulocyte colony-stimulating factor on oral and periodontal
manifestations in a patient with cyclic neutropenia: a case report. Int
J Dent 2009;2009:654239. https://doi.org/10.1155/2009/654239 PMid:20339570 PMCid:PMC2836918
- Welte
K, Zeidler C, Reiter A, et al. Differential effects of
granulocyte-macrophage colony-stimulating factor and granulocyte
colony-stimulating factor in children with severe congenital
neutropenia. Blood 1990; 75:1056-63. https://doi.org/10.1182/blood.V75.5.1056.1056 PMid:1689595
- Boztug
K, Järvinen PM, Salzer E, et al. JAGN1 deficiency causes aberrant
myeloid cell homeostasis and congenital neutropenia. Nat Genet
2014;46:1021-7. https://doi.org/10.1038/ng.3069 PMid:25129144 PMCid:PMC4829076
- Ishida
Y, Higaki A, Tauchi H, et al. Disappearance of neutrophil fluctuations
in a child with cyclic neutropenia by combination therapy of
granulocyte colony-stimulating factor and high-dose immunoglobulin.
Acta Paediatr Jpn 1995;37:388-90. https://doi.org/10.1111/j.1442-200X.1995.tb03338.x PMid:7544058
- Krance
RA, Spruce WE, Forman SJ, et al. Human cyclic neutropenia transferred
by allogeneic bone marrow grafting. Blood 1982;60:1263-6. https://doi.org/10.1182/blood.V60.6.1263.bloodjournal6061263 PMid:6753968
- Rao
S Yao Y, Soares de Brito J, et al. Dissecting ELANE neutropenia
pathogenicity by human HSC gene editing. Cell Stem Cell 2021;28:833-45.
https://doi.org/10.1016/j.stem.2020.12.015 PMid:33513358
- Okada
M, Kobayashi M, Hino T, et al. Clinical periodontal findings and
microflora profiles in children with chronic neutropenia under
supervised oral hygiene. J Periodontol 2001;72:945-52. https://doi.org/10.1902/jop.2001.72.7.945 PMid:11495144
- Zeidler
C, Reiter A, Yakisan E, et al. Long-term treatment with recombinant
human granulocyte colony stimulating factor in patients with severe
congenital neutropenia. Klin Padiatr 1993;205:264-71. https://doi.org/10.1055/s-2007-1025236 PMid:7690865
- Shiohara
M, Shigemura T, Saito S, et al. ELA2 mutations and clinical
manifestations in familial congenital neutropenia. J Pediatr Hematol
Oncol 2009;31:319-24. https://doi.org/10.1097/MPH.0b013e3181984dbe PMid:19415009
[TOP]