A 34-year-old male was admitted to National Cancer Institute, Rio de Janeiro in October 2017 with pancytopenia. The myelogram and BM biopsy showed hypercellularity, myeloid hyperplasia with dysplastic cells and 10% of blasts. The diagnosis was MDS with an excess of blasts-2 (MDS-EB-2) according to WHO classification.[3] The patient was indicated to the treatment with a hypomethylating agent, but this drug was not available at the institution at that time. The patient had blood transfusion support and initiated treatment with recombinant erythropoietin (EPO) combined with folic acid, with no response. In May 2018, the myelogram and BM biopsy showed dysplastic cells and maintained 10% of myeloblasts (Figure 1A-C). Immunophenotyping showed 7.7% of myeloid blast cells expressing medium intensity CD45 and CD117+/HLA-DR+/CD34+/CD38+/CD13+ and dysplastic cells (Figure 1D-H). These results confirmed the previous diagnosis of MDS-EB-2.[3] Cytogenetic analysis of BM cells by G-banding showed: 46,XY,t(11;16)(q23;q24)[5]/46,XY[20] (Figure 1I). Fluorescence in situ hybridization (FISH) was performed using LSI MLL dual color, break apart rearrangement probe (Vysis, Abbott, USA). FISH analysis showed one allele with a split signal indicating the KMT2A-r (Figure 1J). Additionally, the reciprocal translocation was confirmed by using a whole chromosome painting (WCP) probe for chromosome 16 (Vysis, Abbott, USA). No DNMT3A mutations were identified within exons 19, 20, 21, 22, and 23.[4] Allogeneic hematopoietic stem cell transplantation (HSCT) was indicated, but no donor was found. Decitabine was initiated with 20 mg/m2/day intravenous in 1 hour for 5 consecutive days, with cycles of 28/28 days. In November 2018, two months after the fifth cycle of Decitabine, the patient showed improved anemia and thrombocytopenia, although severely neutropenic. In May 2019, the PB count showed: Hb 10 g/dL, platelets 30.000/mm3, 2.000 WBC/mm3, 2% of blasts. The BM immunophenotype showed 5% of myeloid blast cells. The FISH analysis demonstrated KMT2A-r in BM cells. Three months later, immunophenotyping of PB showed 22% of myeloid blast cells, characterizing the evolution from MDS to AML. The patient was hospitalized and underwent remission-inducing chemotherapy for AML (Cytarabine 100 mg/m2/EV/day in continuous infusion for seven consecutive days and Daunorubicin at a dose of 60 mg/m2/day EV on days D1/D2/D3). In February 2020, he was submitted to intensification chemotherapy [Cytarabine 1.5g/m2/EV/12/12-hours on days D1/D3 and D5, totaling six doses of Cytarabine (2nd intensification cycle)]. No blast cells were observed at PB, but the patient evolved with gradual severe cytopenias. Until April 2020, there was no BM recovery. In June 2020, blood counts revealed an expansion of the leukemic clone, presenting more than 50% of blasts in PB. Treatment with subcutaneous Cytarabine was initiated (200 to 300 mg/week), with no hematological response. Despite all efforts, the patient presented bleeding from the central nervous system in August 2020, secondary to treatment-refractory hyperleukocytosis and central leukocytosis, evolving to death. The summary of the steps showing the evolution from MDS to AML is described in Figure 2.
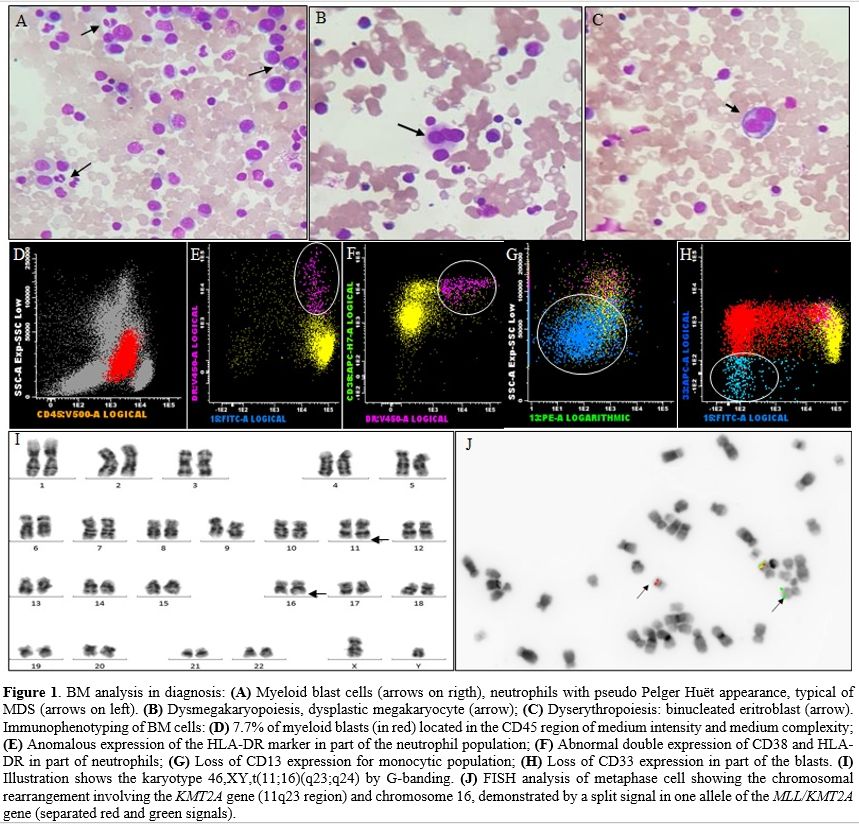 |
Figure 1. BM analysis in diagnosis: (A) Myeloid blast cells (arrows on rigth), neutrophils with pseudo Pelger Huët appearance, typical of MDS (arrows on left). (B) Dysmegakaryopoiesis, dysplastic megakaryocyte (arrow); (C) Dyserythropoiesis: binucleated eritroblast (arrow). Immunophenotyping of BM cells: (D) 7.7% of myeloid blasts (in red) located in the CD45 region of medium intensity and medium complexity; (E) Anomalous expression of the HLA-DR marker in part of the neutrophil population; (F) Abnormal double expression of CD38 and HLA-DR in part of neutrophils; (G) Loss of CD13 expression for monocytic population; (H) Loss of CD33 expression in part of the blasts. (I) Illustration shows the karyotype 46,XY,t(11;16)(q23;q24) by G-banding. (J) FISH analysis of metaphase cell showing the chromosomal rearrangement involving the KMT2A gene (11q23 region) and chromosome 16, demonstrated by a split signal in one allele of the MLL/KMT2A gene (separated red and green signals). |
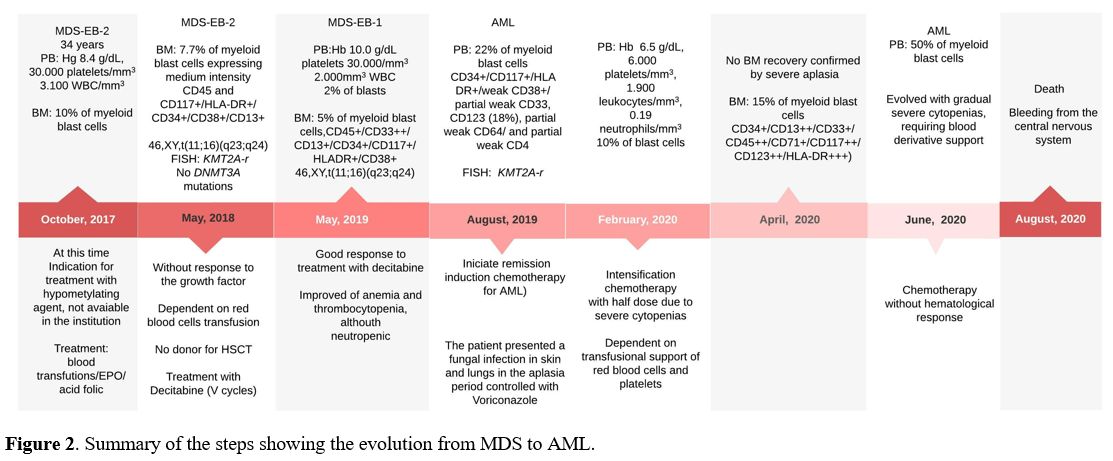 |
Figure 2. Summary of the steps showing the evolution from MDS to AML. |
In MDS, chromosomal translocations as sole chromosomal abnormality involving chromosome 11 occur in approximately 0.2% of patients.[1] Due to the low number of patients showing 11q23/KMT2A translocations in MDS, their real prognostic impact is unknown, and patients with this genetic alteration are assigned to the intermediate-risk group in IPSS-R.[1,2] Our report describes the clinical outcome in a young adult patient with de novo MDS showing t(11;16)(q23;q24) with KMT2A-r. Considering the age of our patient, some studies in MDS (mainly associated with HSCT) had, in their cohort, patients between 18 and 55 years, therefore including young adult patients.[5] In our patient, the diagnosis of MDS-EB-2 was made according to the criteria of WHO classification.[3] The immunophenotyping analysis showed 7.7% of blasts and dysplastic features in the neutrophil population, monocytic and loss of CD33 in blast cells, characteristics not observed in AML, even with a slower dynamics course.[3] A broad scientific review showed that only four cases of acute leukemia with t(11;16)(q23;q24) had been described so far.[6-9] The patients described with t(11;16)(q23;q24) were associated with leukemia relapse and refractoriness to treatment. In the present study, the patient also had a poor clinical outcome, progressing from MDS to AML and refractoriness to treatment. It is important to note that the hypomethylating agents(HMA) have been considered the standard of care for MDS patients. However, HMA is not curative. Response to these drugs occurs in approximately 50% of patients, and the duration of response is transient because HMAs do not eradicate neoplastic clones.[10] HSCT remains the only possible curative option for MDS patients.[5,10] This study and literature review highlight the importance of cytogenetics, molecular tests, and clinical follow-up of de novo MDS patients to identify new prognostic risk groups and research new therapeutic drugs for these patients. Since this study was the first that described the t(11;16) with KMT2A-r associated with AML evolution and poor prognosis in a patient with MDS, it is necessary new studies involving a more number of patients to provide a better understanding of t(11;16) with KMT2A-r involved in MDS pathogenesis and its prognosis impact.