Paul Kambale-Kombi1, Roland Marini Djang’eing’a3,4, Jean-Pierre Alworong’a Opara2, Jean-Paulin Mbo Mukonkole1, Vincent Bours5, Serge Tonen-Wolyec1, Dieu-Merci Mbumba Lupaka1, Lucien Bolukaoto Bome1, Charles Kayembe Tshilumba1 and Salomon Batina-Agasa1..
1 Department
of Internal Medicine, Faculty of Medicine and Pharmacy, University of
Kisangani, Kisangani, Democratic Republic of the Congo.
2
Department of Pediatrics, Faculty of Medicine and Pharmacy, University
of Kisangani, Kisangani, Democratic Republic of the Congo.
3 Department of Pharmacy, Faculty of Medicine and Pharmacy, University of Kisangani, Kisangani, Democratic Republic of the Congo.
4
Department of Pharmaceutical Sciences, Laboratory of Analytical
Chemistry, Faculty of Medicine, University of Liège, Liège, Belgium.
5 Department of Human Genetics, University Hospital of Liège, Faculty of Medicine, University of Liège, Liège, Belgium.
Correspondence to:
Paul Kambale-Kombi, Department of Internal Medicine, Faculty of
Medicine and Pharmacy, University of Kisangani, Kisangani, Democratic
Republic of the Congo. E-mail:
kombikambalepaul@gmail.com
Published: July 1, 2022
Received: November 19, 2021
Accepted: June 3, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022046 DOI
10.4084/MJHID.2022.046
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and objective:
Sickle cell disease (SCD) is now a well-established cause of renal
damage. In the northeast of the Democratic Republic of Congo (DRC), SCD
is common. However, sickle cell nephropathy remains unstudied in this
region. Thus, this study aimed to assess renal abnormalities in SCD
patients in Kisangani (northeastern DRC).
Methods:
This cross-sectional study included 98 sickle cell patients selected
from six health facilities in Kisangani and 89 healthy non-sickle cell
subjects as the control group. Based on a survey form, a clinical
examination and biological tests were performed to collect data related
to the sex, age, weight, height, pressure, serum creatinine, serum uric
acid, urinary albumin/creatinine ratio, and hemoglobin phenotype. We
used a spectrophotometer to measure serum creatinine and uricemia, the
sickle SCAN® device for hemoglobin phenotype, and an automatic
multifunction analyzer for urine albumin/creatinine ratio. Data were
entered into an Excel file and analyzed on SPSS 20.0.
Results:
The mean urine albumin-to-creatinine ratio was 11.79±9.03 mg/mmol in
SCD patients, significantly higher than in AA (1.69±1.89 mg/mmol) and
AS (2.97±4.46 mg/mmol) subjects. The decrease in glomerular filtration
rate was more observed in SCD patients with hyperuricemia compared to
those with normal uric acid levels. A significantly elevated prevalence
of chronic kidney disease was observed among SCD patients (87.8%)
compared to 23.8% in AS and 7.7% in AA subjects.
Conclusions:
This study highlighted that albuminuria and chronic kidney disease are
common in SCD patients in Kisangani. More studies are needed to further
document these complications.
|
Introduction
Sickle
cell disease (SCD) is now a well-established cause of renal damage
associated with high mortality in SCD patients. Renal involvement
contributes to the diminished life expectancy of patients with SCD,
accounting for 16–18% of mortality.[1] In the study by Hamideh and Alvarez[2]
in the United States of America, nephropathy accounted for 16,4% of
deaths in children and adults with SCD. Therefore, detecting renal
abnormalities at an early stage when interventions may be more
effective is essential to improve the lifespan of SCD patients.
Sickle
cell disease patients may present with several types of renal
dysfunction. Different pathophysiologic mechanisms have been proposed
to explain the development of sickle cell nephropathy, where
hemolysis-related vasculopathy[3] and vaso-occlusive phenomena[4]
are the main contributors. Glomerular hyperfiltration and
microalbuminuria/proteinuria are early manifestations of sickle cell
nephropathy. Hyperfiltration starts as early as 9–19 months.[5] Over time, hyperfiltration may lead to microalbuminuria and later to renal failure.
Aygun et al.[6]
demonstrated that hydroxyurea treatment, which is currently the only
substantive treatment for SCD, was associated with improved renal
function in SCD patients presenting hyperfiltration. However, few
patients have access to hydroxyurea treatment in sub-Saharan Africa,
including the Democratic Republic of the Congo (DRC). In general, there
is still poor utilization of standard-care practices for SCD patients,[7-9] and that exposes SCD patients living in this area to the risk of multi-organ damage. In Ghana, Anto et al.[10]
reported chronic renal disease related to sickle cell anemia in 73.5%
of patients under 13 years of age. In Kinshasa (western part of the
DRC), Aloni et al[8] reported a high prevalence of
hyperfiltration among children with SCD, approximately one in three
children. However, this study included only children younger than 14
years and moreover, in a center dedicated to the management of SCD and
a teaching hospital. A study on adolescent and adult subjects with SCD
has been made in Nigeria, where 204 HbSS patients were recruited.[11]
The prevalence of chronic kidney disease (CKD) was 38.9%. eGFR
(Estimated Glomerular Filtration Rate) showed that 69 (26.8%) had
hyperfiltration; 35 (13.6%) stage 1 CKD; 53 (20.6%) stage 2 CKD; 33
(12.8%) stage 3a CKD; 28 (10.9%) stage 3b CKD; 30 (11.7%) stage 4 CKD
and 9 (3.5%) had end-stage renal disease; predictors of CKD using eGFR
include age, Systolic Blood Pressure, number of units of blood
transfusion, Packed red cell, urea, creatinine, and uric acid levels.[11]
In
the northeast of the DRC, this issue remains unstudied. However, given
the context of poor management of SCD as reported by Kambale et al.[7]
in this region, renal abnormalities may be of greater magnitude.
Therefore, this study aimed to evaluate renal abnormalities in subjects
with SCD in Kisangani (northeastern DRC), a city located about 1250
kilometers from Kinshasa and in one of three regions where
approximately one-third of sickle cell births in the DRC occur.[12]
Methods
Context and ethical considerations.
This study was carried out in Kisangani from January to May 2020 with
the approval of the Ethical Committee of the University of Kisangani
(Réf. UNIKIS/CER/007/2018). In addition, we obtained authorization from
the Tshopo Provincial Health Division to survey the health facilities
of the Tshopo province (N°701/DPS/TSHOPO/SEC/019/2019). Enrollment in
the study was conditional on free and informed consent from
participants. For individuals under 18 years of age, parental and/or
guardian consent was required. The objectives and purpose of the study
were explained, and subjects were informed of their right to refuse to
participate or to withdraw from it at any time without this having any
implication on their care. Confidentiality of results was guaranteed by
ensuring anonymity in the data processing.
Study design and settings.
This care facility-based survey was a cross-sectional study conducted
in six health facilities in the city of Kisangani (Cliniques
universitaires de Kisangani, Hôpital général de référence
Makiso/Kisangani, Hôpital général de référence de Kabondo, Centre de
santé ALABUL, Centre d’anémie SS «Gracia fondation», Cliniques
Stanley), selected on the basis of their high attendance by SCD
patients. Kisangani is one of the third largest cities in the DRC with
an estimated population of 1.6 million inhabitants,[13] the neonatal prevalence of homozygous form of SCD (SS) about 1%; and that of heterozygous form (AS) of 23.3%.[14]
In addition, it is an area where insufficient use of standard care
practices for SCD patients has been recently reported (2021).[7]
Study population and sampling. The study population consisted of homozygous SCD patients (HbSS) previously diagnosed by the sickle SCAN® rapid test[15]
and followed up from January to May 2020 in the health facilities
mentioned above. They were included and surveyed during their follow-up
appointment according to an exhaustive sampling. The control group
consisted of healthy individuals with no SCD siblings of SCD patients,
selected by non-probability convenience sampling. For their enrollment,
sickle cell patients and/or their parents/guardians were sensitized and
informed about the importance of screening for SCD and its renal
complications during follow-up appointments. They, in turn, sensitized
their family members. Family members who agreed to be screened came to
the selected health facilities to be surveyed. The same inclusion and
exclusion criteria were used for the study population (sickle cell
disease patients and control group).
Inclusion criteria were:
(1) being at least five years old, (2) having given consent to
participate in the study or parental/guardian consent for subjects
under 18 years of age, and (3) not having received a blood transfusion
for at least three months. In addition, for sickle cell disease
patients, (4) to be in inter critical stage, defined as the absence of
acute complications of SCD or signs of other diseases for at least two
weeks.
Exclusion criteria were:
having a history of (1) hypertension, (2) diabetes mellitus, (3)
frequent use of traditional therapy, (4) having signs of urinary tract
infection on urinary sediment analysis or signs of other diseases at
the time of the survey. In addition, for sickle cell disease patients,
(5) being under hydroxyurea treatment.
Data collection.
A survey form was used to collect data from the study participants. The
following clinical and laboratory data were collected: (1) demographic
and anthropometric characteristics [gender, age, weight, height], (2)
Blood pressure, (3) serum creatinine, (4) serum uric acid, (5) urine
albumin/creatinine ratio and (6) hemoglobin phenotype.
A physical
examination collected the clinical data. Blood pressure was measured
using standard procedure. Next, the weight and the height were measured
using the weighing scale Seca® and stadiometer Seca®, respectively.
Finally, the axillary temperature was measured by a mercury
thermometer.
Sample collection and laboratory analysis.
Blood collection.
For each subject, 3 mL of whole blood was collected by vein puncture
into a vacutainer tube with EDTA and 2 mL into a vacutainer tube
without anticoagulant for the serum creatinine and uric acid.
Following
vein puncture, samples were stored at 4–8°C in an isothermal box
containing cold packs and then transferred to the laboratory within two
hours at most. The serum uric acid and serum creatinine levels were
estimated enzymatically by a spectrophotometer using commercial Uric
Acid and Creatinine Assay kits from Cypress Diagnostics (Cypress
Diagnostics: Nijverheidsstraat 8.2235 Hulshout. Belgium) following the
procedure outlined in the manuals supplied with the kits. According to
the manufacturer's instructions, hemoglobin phenotyping was performed
using the sickle SCAN® rapid test.[15]
Urine collection. The urine of each participant was collected into a clean, sterile, and leak-proof container.
The urine albumin/creatinine ratio was determined using an automatic
multifunction analyzer (Model: Icare-2100, Changsha Sinocare
Inc.NO.265, Guyuan Road, Hi-tech Zone, Changsha, Hunan Province 410205,
China). In addition, a drop of decanted urine deposit was observed
under a microscope for urine cytobacteriological examination.
Outcomes criteria: Estimated glomerular filtration rate (eGFR) and chronic kidney disease.
For the subjects under 18 years of age, the updated Schwartz equation[16]
was used for calculating the eGFR. For subjects aged 18 years and
older, we used the MDRD (Modification of Diet in Renal Disease)[17] formula.
Chronic kidney disease (CKD) was classified according to the Kidney Disease Improving Global Outcome (KDIGO)[18] as follow:
Stage 1: ≥ 90 ml/min/1.73 m2 (kidney damage with normal or increased eGFR);
Stage2: 60–89 ml/min/ 1.73m2 (kidney damage with mildly decreased eGFR);
Stage 3a: 45–59 ml/min/1.73 m2 (mildly to moderately decreased eGFR);
Stage 3b: 30–44 ml/min/1.73 m2 (moderate to severely decreased eGFR);
Stage 4: 15–29 ml/min/1.73 m2 (severely decreased eGFR) and
Stage 5: <15 ml/min/1.73 m2 (Kidney failure).
The
urine albumin-to-creatinine ratio scored kidney damage≥ 3 mg/mmol;
hyperfiltration was defined as an eGFR greater than 140 ml/min/1.73 m2.
Subjects were considered hyperuricemia if their total serum uric acid
concentrations were greater than the upper limits of normal for age and
sex.
Statistical Analysis.
The data were entered into an electronic database (Microsoft Excel
2019), and all the analyzes were carried out on statistical software
(SPSS 20.0). The quantitative variables were compared using the mean
and its standard deviation. The qualitative variables were described
with the numbers and proportions of each modality. An analysis of the
variance(ANOVA) was used to compare a quantitative variable between
hemoglobin phenotype subgroups. In the case of heterogeneity of data
variances, we used Welch homogeneity correction. For the crossing
between several qualitative variables, the Chi2
test was used if the application conditions allowed it. If not,
Fisher's exact test was performed. The risk of species 1 alpha was set
at 5% for all analyzes, and Significant levels were measured at 95% CI.
Results
The
study was conducted on 98 SCD patients and 89 no sickle cell subjects
as the control group (26 AA and 63 AS subjects). The procedure of
participant recruitment is outlined in Figure 1.
There were 51 (52%) females and 47 (48%) males in the SCD patients; 50
(56.2%) females and 39 (43.8) males in the control group without
significant difference. The mean age of SCD patients was 12.82±7.5215
years, and that of AA and AS was 15.54±6.65 years and 12.71±7.68 years,
respectively. Overall, hyperuricemia was observed in 39.8% of SCD
patients versus 10.1% in the control group (Table 1).
Furthermore, the mean uric acid level was significantly higher in SCD
patients compared to AA (p<0.01) and AS (p<0.001) subjects (Figure 2).
In addition, the decrease in eGFR was more observed in SCD patients
with hyperuricemia than SCD patients with normal uric acid levels.
However, there was no significant difference (Figure 3).
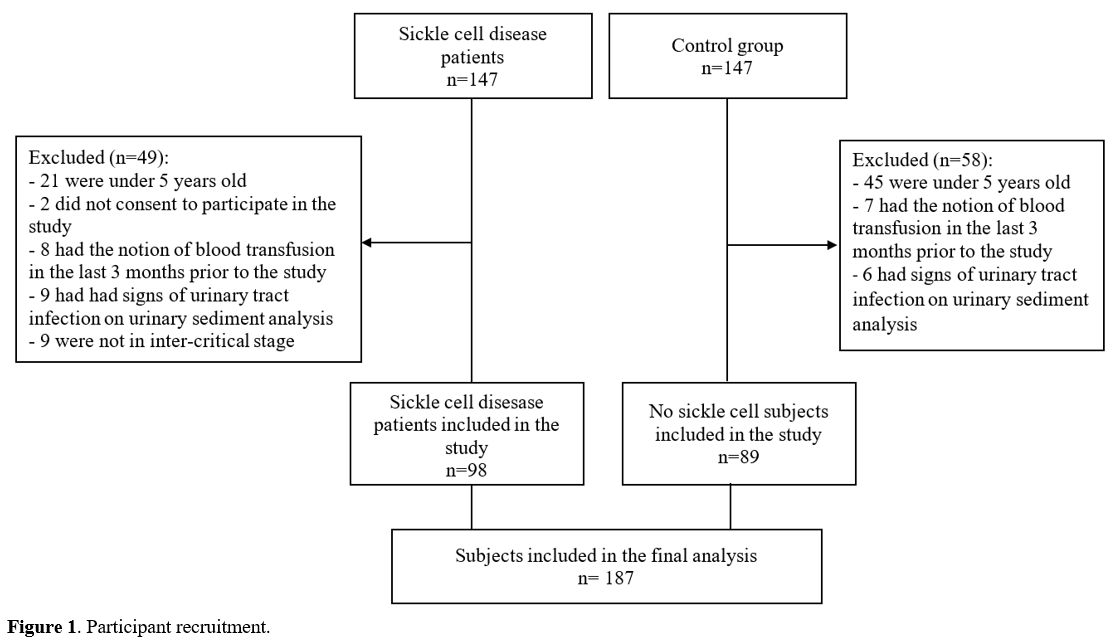 |
Figure 1. Participant recruitment. |
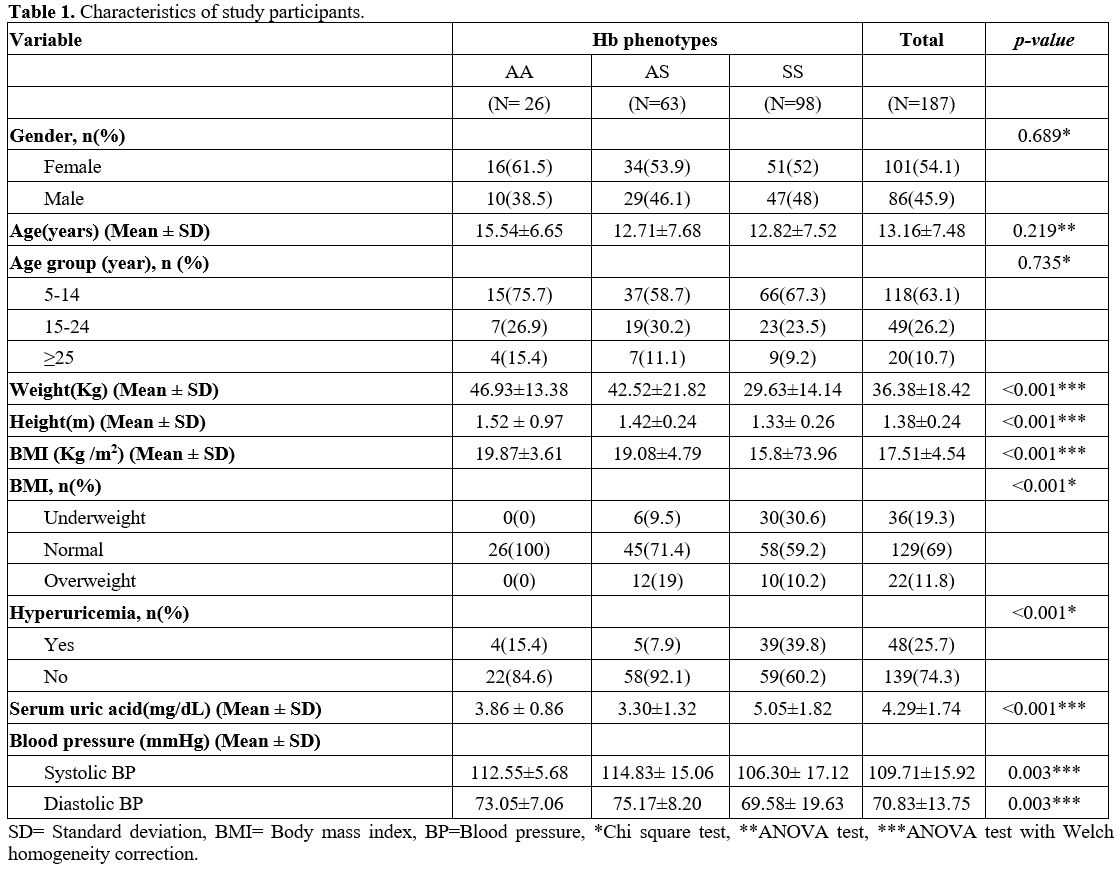 |
Table
1. Characteristics of study participants. |
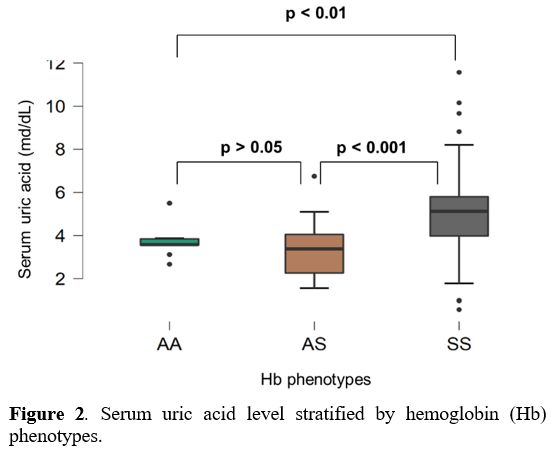 |
Figure 2. Serum uric acid level stratified by hemoglobin (Hb) phenotypes. |
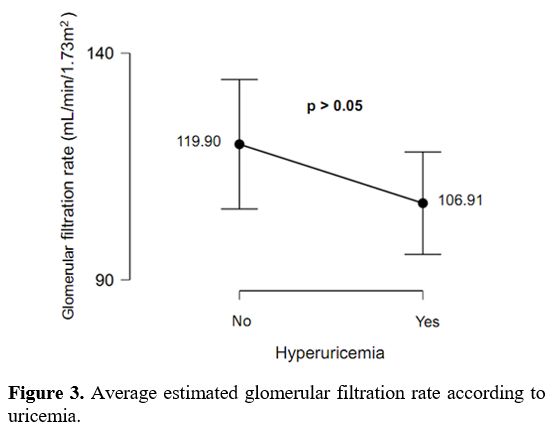 |
Figure 3. Average estimated glomerular filtration rate according to uricemia. |
Microalbuminuria
was more prevalent in SCD patients, 85 subjects (86.7%) versus 21
subjects (23.6%) in the control group. In addition, the mean urine
albumin-to-creatinine ratio was 11.79±9.03 mg/mmol in SCD patients,
significantly higher than in AA (1.69±1.89 mg/mmol) and AS (2.97±4.46
mg/mmol) subjects. Hyperfiltration was observed in 22.4% of the SCD
patients, whereas no case was observed in the control group (Table 2).
Compared to AA (66.6 µmol/L ± 8.52 µmol/L) and AS (70.71 µmol/L ± 17.12
µmol/L) subjects, SCD patients had significantly lower mean serum
creatinine levels (52.59 µmol/L±18.59 µmol/L) (p < 0.001) (Figure 4).
A significantly elevated prevalence of CKD disease was observed among
SCD patients (87.8%) compared to 23.8% and 7.7% observed in AS and AA
subjects, respectively. Of the 87.8% of SCD patients with CKD, 83.7%
were in stages 1 and 2 of the disease, and 5.3% were in stage 3a (Table 2).
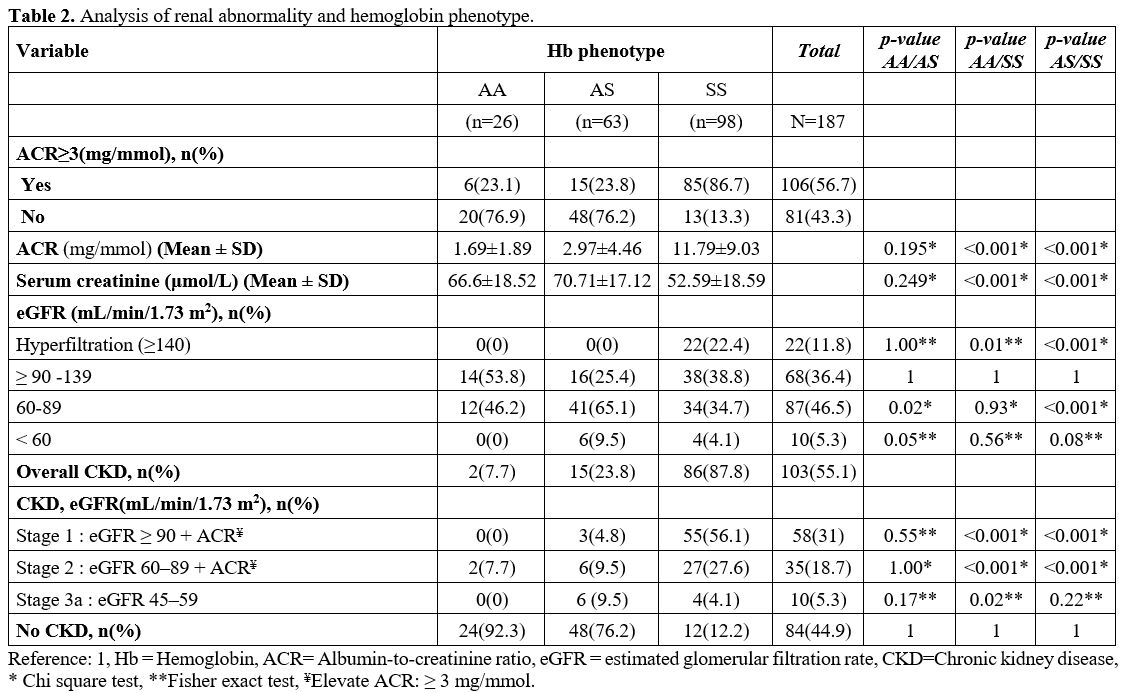 |
Table 2. Analysis of renal abnormality and hemoglobin phenotype. |
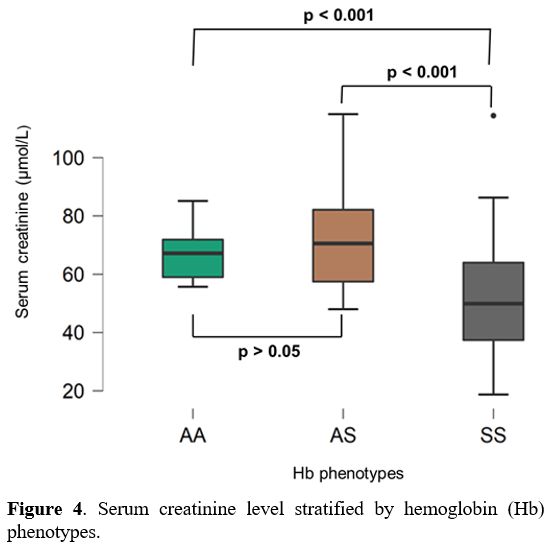 |
Figure 4. Serum creatinine level stratified by hemoglobin (Hb) phenotypes. |
Discussion
This
study reported the abnormalities of uricemia and renal function
observed in SCD patients in Kisangani, where SCD is still undermanaged.
Overall, the results showed that hyperuricemia was observed in 39.8% of
SCD patients versus 10.1% in the control group. The decrease in eGFR
was more in SCD patients with hyperuricemia than in those with normal
uric acid levels. Microalbuminuria was more prevalent in SCD patients
(86.7%) compared to the control group (23.6%). The mean
albumin-to-creatinine ratio was 11.79±9.03 mg/mmol in SCD patients,
significantly higher than in AA and AS subjects. CKD prevalence in SCD
patients was 87.8%, considerably higher than the 23.8% and 7.7%
observed in AS and AA subjects.
Sickle cell patients have an
increased risk of developing chronic kidney disease, primarily because
of the renal pathophysiology associated with chronic heme exposure and
sickling of red blood cells. However, other factors such as
hyperuricemia and hypertension, which are established modifiable risk
factors associated with the development of CKD in no SCD populations,
could play a role in the occurrence of this complication.[19]
This study observed hyperuricemia in 39.8% of SCD patients versus 10.1%
of controls. The mechanism of the development of hyperuricemia is
either uric acid overproduction or inefficient renal excretion. In SCD,
the mechanism often suggested is overproduction from an increased
marrow activity and turnover of nucleic acids secondary to hemolysis.
Several studies have reported elevated uric acid levels in SCD
patients: 26.9%,[20] 30%,[21] 28%,[22] 9.2%.[23]
Compared to these studies, the higher prevalence of hyperuricemia
observed in this study could be explained, on the one hand, by the
hemolytic phenomena related to the endemicity of infectious and
parasitic diseases in our environment. On the other hand, it could be
explained by the poor management of SCD in Kisangani, as reported by
Kambale et al.,[7] which exposes patients to chronic
hemolysis. Preventive measures, effective management of infections, and
hydroxyurea could reduce the frequency of hyperuricemia. Normalization
of serum uric acid levels in SCD patients is of great importance since,
in this study, the decrease in eGFR was more observed in SCD patients
with hyperuricemia (average eGFR = 106.91± 34.81 mL/min/1.73m2) compared to SCD patients with normal uric acid levels (average eGFR = 119.90 ± 53.78 mL/min/1.73m2)
(p=0.512) (p>0.05). Although the difference was not statistically
significant, these results suggest a possible link between
hyperuricemia and eGFR decrease in patients with SCD. Thus, our
observation is in agreement with that of Kaspar et al., who reported a
decrease in mean eGFR in SCD patients with hyperuricemia compared to
those with normal uric acid levels.[24] Similarly, in
Lebensburger et al., SCD patients with hyperuricemia showed a
significant decrease in mean eGFR compared to those without
hyperuricemia.[19] In addition, hyperuricemia would
be involved in certain painful crises observed in sickle cell patients.
Although there is still little evidence supporting this hypothesis,
Gupta et al. reported that in SCD patients, not all pain is sickle cell
pain.[25] Therefore, these findings further buttress
the need to improve the management of SCD in the DRC. Nevertheless,
further studies are strongly needed because if there is evidence for a
link between hyperuricemia and renal dysfunction on the one hand and
hyperuricemia and painful crises in SCD patients on the other,
hyperuricemia will be a potential therapeutic target.
The assessment of renal function in SCD patients has been the subject of numerous studies.[10,26-32]
However, these studies have been conducted with different exploration
methods, on diverse populations, and under different management
settings. These different methods have led to widely varying results,
with a prevalence of sickle cell nephropathy ranging from less than 10%
to values as high as 73.5%, depending on the above factors and the
criteria retained in the definition of renal dysfunction.
The
most commonly considered markers of sickle cell nephropathy are
albuminuria, hyperfiltration, and CKD. In this study, the albuminuria,
evaluated by the urine albumin-to-creatinine ratio, was more prevalent
in SCD patients [85 of 98 subjects (86.7%) vs. 21 of 89 issues (23.6%)
in controls]. In addition, CKD was present in 87.8% of SCD patients vs.
19.1% of controls. The prevalence of albuminuria resulting from sickle
cell nephropathy is high, varying between 26 and 68% in adult patients.[26] However, consistent with our results, a higher prevalence of CKD has been reported by Anto et al. (73.5%),[10] Isaza-López et al. (70%),[27] and Ephraim et al. (68,4 %).[28]
Nevertheless, the prevalence observed in our study is higher than that
reported elsewhere, and various factors, including the age of the
respondents, the quality of SCD management, the infectious context, the
haplotype found in the Democratic Republic of the Congo, and the method
of assessing proteinuria may account for this. Indeed, the studies that
reported lower prevalences of sickle cell nephropathy were mostly
conducted in children.[29,32,33] According to Gosmanova et al.[34] and Adebayo et al.,[35]
the Clinical presentation of sickle cell nephropathy is age-dependent,
with kidney dysfunction slowly beginning to develop from childhood,
progressing to CKD and kidney failure during the third and fourth
decades of life.[11] In SCD patients, Ranque et al.
observed sickle cell nephropathy in 27% of children younger than 10
years, and its frequency increased with age (48% in SCD patients aged
>40 years).[30] Therefore, our result could also be explained because our study included both children and adults.
The
beneficial effect of hydroxyurea on sickle cell nephropathy has been
demonstrated by Laurin et al., who observed proteinuria in 34.7% of SCD
patients receiving hydroxyurea versus 55.4% among those not receiving
this medication.[36] However, in the Democratic
Republic of the Congo, one of the major challenges in managing SCD is
the accessibility of hydroxyurea to patients. The study by Kambale et
al. showed that in the northeastern part of this country, only 5.1% of
SCD patients have ever been treated with hydroxyurea,[7] and this is a condition that can promote sickle cell nephropathy.
Different
pathophysiological mechanisms have been proposed to explain the
development of sickle cell nephropathy, where hemolysis and vascular
occlusion are the main contributors.[4] In the
Kisangani setting, characterized by the endemicity of infectious and
parasitic diseases and the poor management of SCD as documented in the
milieu,[7] hemolysis and vaso occlusion could be
frequent and account for the high prevalence reported in this study. We
also speculate that the Bantu haplotype, the most common in the
Democratic Republic of the Congo and associated with severe disease
manifestations, may be an additional factor in the development of
sickle cell nephropathy in the Democratic Republic of the Congo.
Finally,
due to the COVID-19 pandemic, the albumin-to-creatinine ratio was used
for proteinuria evaluation without a second confirmation test. Thus,
the possibility of false-positive results is a limitation for this
study since high transient albuminuria has been reported by Kim et al.[37]
Limitations
As
mentioned above, the urine albumin/creatinine ratio was performed to
assess sickle cell nephropathy in the resource-limited setting of
Kisangani (DRC), and the occurrence of the Covid-19 pandemic.
Therefore, the urine albumin/creatinine ratio was performed once
without repetition of tests to confirm the abnormalities. This
limitation of the study implies that the high prevalence of albuminuria
and CKD observed may be overestimated. Hence, we have interpreted the
findings with caution. Nevertheless, despite this limitation, the
results of this study globally indicate that sickle cell nephropathy is
very common in northeastern DRC.
Conclusions
This
study highlighted a high prevalence of hyperuricemia, albuminuria, and
chronic kidney disease in SCD patients in northeastern DRC. Therefore,
current management should routinely screen for and address renal
complications, which could likely contribute to decreased morbidity and
mortality of this disease. In addition, studies evaluating the benefit
of angiotensin-converting enzyme inhibitors or angiotensin II receptor
blockers in the management of sickle cell nephropathy are strongly
required in the DRC. Similarly, the link between hyperuricemia and
sickle cell nephropathy should be further studied. Finally, there is an
urgent need to implement dedicated centers offering comprehensive care
to SCD patients in the DRC.
Acknowledgments
The authors are grateful to Judith Tibamwenda Akiki for her participation in the data collection. They also thank the Belgian Académie de Recherche et d’Enseignement Supérieur (ARES) for the PhD scholarship.
References
- Nath KA, Hebbel RP. SCD: renal manifestations and mechanisms. Nat Rev Nephrol. 2015;11(3):161-171. doi:10.1038/nrneph.2015.8 https://doi.org/10.1038/nrneph.2015.8 PMid:25668001 PMCid:PMC4701210
- Hamideh
D, Alvarez O. SCD related mortality in the United States (1999-2009).
Pediatr Blood Cancer. 2013;60(9):1482-1486. doi:10.1002/pbc.24557 https://doi.org/10.1002/pbc.24557 PMid:23637037
- Kato
GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the
pathophysiology of SCD. J Clin Invest. 2017;127(3):750-760.
doi:10.1172/JCI89741 https://doi.org/10.1172/JCI89741 PMid:28248201 PMCid:PMC5330745
- Hariri
E, Mansour A, El Alam A, Daaboul Y, Korjian S, Aoun Bahous S. Sickle
cell nephropathy: an update on pathophysiology, diagnosis, and
treatment. Int Urol Nephrol. 2018;50(6):1075-1083.
doi:10.1007/s11255-018-1803-3 https://doi.org/10.1007/s11255-018-1803-3 PMid:29383580
- Ware
RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, Shulkin BL,
Shalaby-Rana E, Strife CF, Miller JH, Lane PA, Wang WC, Miller ST, BABY
HUG Investigators. BABY HUG Investigators. Renal function in infants
with sickle cell anemia: baseline data from the BABY HUG trial. J
Pediatr. 2010; 156(1):66-70. https://doi.org/10.1016/j.jpeds.2009.06.060 PMid:19880138 PMCid:PMC4755353
- Aygun
B, Mortier NA, Smeltzer MP, Shulkin BL, Hankins JS, Ware RE.
Hydroxyurea treatment decreases glomerular hyperfiltration in children
with sickle cell anemia. Am J Hematol. 2013;88(2):116-119.
doi:10.1002/ajh.23365 https://doi.org/10.1002/ajh.23365 PMid:23255310 PMCid:PMC4673980
- Kambale-Kombi
P, Marini Djang'eing'a R, Alworong'a Opara JP, Minon JM, Boemer F,
Bours V, Tonen-Wolyec S, Kayembe Tshilumba C, Batina-Agasa S.
Management of SCD: current practices and challenges in a northeastern
region of the Democratic Republic of the Congo. Hematology.
2021;26(1):199-205. doi:10.1080/16078454.2021.1880752 https://doi.org/10.1080/16078454.2021.1880752 PMid:33594960
- Aloni
MN, Nkee L. Challenge of managing SCD in a pediatric population living
in Kinshasa, democratic republic of congo: a sickle cell center
experience. Hemoglobin. 2014;38(3):196-200.
doi:10.3109/03630269.2014.896810 https://doi.org/10.3109/03630269.2014.896810 PMid:24669956
- Ofakunrin
A, Oguche S, Adekola K, Okpe ES, Afolaranmi TO, Diaku-Akinwumi IN,
Zoakah AI, Sagay AS. Effectiveness and Safety of Hydroxyurea in the
Treatment of Sickle Cell Anaemia Children in Jos, North Central
Nigeria. J Trop Pediatr. 2020;66(3):290‐298. https://doi.org/10.1093/tropej/fmz070 PMid:31608959 PMCid:PMC7249733
- Anto
EO, Obirikorang C, Acheampong E, Adua E, Donkor S, Afranie BO, Ofori M,
Asiamah EA, Adu EA. Renal abnormalities among children with sickle cell
conditions in highly resource-limited setting in Ghana. PLoS One.
2019;14(11):e0225310.
https://doi.org/10.1371/journal.pone.0225310 PMid:31743364 PMCid:PMC6863548 - Bukar
AA, Sulaiman MM, Ladu AI, Abba AM, Ahmed MK, Marama GT, Abjah UM.
Chronic Kidney Disease amongst Sickle Cell Anaemia Patients at the
University of Maiduguri Teaching Hospital, Northeastern Nigeria: A
Study of Prevalence and Risk Factors. Mediterr J Hematol Infect Dis.
2019 Jan 1;11(1):e2019010. doi: 10.4084/MJHID.2019.010. https://doi.org/10.4084/mjhid.2019.010 PMid:30671216 PMCid:PMC6328039
- Piel
FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH,
Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle
haemoglobin in neonates: a contemporary geostatistical model-based map
and population estimates. Lancet. 2013;381(9861):142-151.
doi:10.1016/S0140-6736(12)61229-X https://doi.org/10.1016/S0140-6736(12)61229-X
- Tonen-Wolyec
S, Batina-Agasa S, Muwonga J, Mboumba Bouassa RS, Kayembe Tshilumba C,
Bélec L. Acceptability, feasibility, and individual preferences of
blood-based HIV self-testing in a population-based sample of
adolescents in Kisangani, Democratic Republic of the Congo. PLoS One.
2019;14(7):e0218795. https://doi.org/10.1371/journal.pone.0218795 PMid:31260480 PMCid:PMC6602204
- Agasa
B, Bosunga K, Opara A, Tshilumba K, Dupont E, Vertongen F, Cotton F,
Gulbis B. Prevalence of SCD in a northeastern region of the Democratic
Republic of Congo: what impact on transfusion policy?. Transfus Med.
2010;20(1):62-65. doi:10.1111/j.1365-148.2009.00943.x https://doi.org/10.1111/j.1365-3148.2009.00943.x PMid:19712051
- Segbena
AY, Guindo A, Buono R, Kueviakoe I, Diallo DA, Guernec G, Yerima M,
Guindo P, Lauressergues E, Mondeilh A, Picot V, Leroy V. Diagnostic
accuracy in field conditions of the sickle SCAN® rapid test for SCD
among children and adults in two West African settings: the DREPATEST
study. BMC Hematol. 2018;18:26. Published 2018 Sep 17.
doi:10.1186/s12878-018-0120-5 https://doi.org/10.1186/s12878-018-0120-5 PMid:30237894 PMCid:PMC6142627
- Mian
AN, Schwartz GJ. Measurement and Estimation of Glomerular Filtration
Rate in Children. Adv Chronic Kidney Dis. 2017;24(6):348-356.
doi:10.1053/j.ackd.2017.09.011 https://doi.org/10.1053/j.ackd.2017.09.011 PMid:29229165 PMCid:PMC6198668
- Jha
V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang
AY, Yang CW. Chronic kidney disease: global dimension and perspectives
[published correction appears in Lancet. 2013Jul20;382(9888):208].
Lancet.2013;382(9888):260-272. doi:10.1016/S0140-6736(13)60687-X https://doi.org/10.1016/S0140-6736(13)60687-X
- Summary of Recommendation Statements. Kidney Int Suppl (2011). 2013;3(1):5-14. doi:10.1038/kisup.2012.77 https://doi.org/10.1038/kisup.2012.77 PMid:25598998 PMCid:PMC4284512
- Lebensburger
JD, Aban I, Hilliard LM, Feig DI. Hyperuricemia and abnormal nocturnal
dipping impact glomerular filtration rate in patients with sickle cell
anemia. Am J Hematol. 2021;96(5):E143-E146. doi:10.1002/ajh.26115 https://doi.org/10.1002/ajh.26115 PMid:33524174
- Akintayo
R, Adelowo O, Chijioke A, Olanrewaju T, Olufemi-Aworinde K, Akintayo F.
Gout Is More Frequent in SCD Than the General Population. Arthritis
Rheumatol.2017; 69 (suppl 10). https://acrabstracts.org/abstract/gout-is-more-frequent-in-sickle-cell-disease-than-the-general-population/. Accessed October 19, 2021
- Lebensburger
JD, Cutter GR, Howard TH, Muntner P, Feig DI. Evaluating risk factors
for chronic kidney disease in pediatric patients with sickle cell
anemia. Pediatr Nephrol. 2017;32(9):1565-1573.
doi:10.1007/s00467-017-3658-8 https://doi.org/10.1007/s00467-017-3658-8 PMid:28382567 PMCid:PMC5628098
- Matar
K, Moustapha D, Kesso B, Ngor S, Mareme T, Mame M, Malick N, Fatou G,
Philomene L, Aynina C, Amadou D, Madieye G. Association between Uric
Acid and Metabolic Syndrome in Homozygous Sickle Cell Patients.
Advances in Biological Chemistry 202;11:142-148.
doi:10.4236/abc.2021.113010. https://doi.org/10.4236/abc.2021.113010
- Arlet
JB, Ribeil JA, Chatellier G, Pouchot J, de Montalembert M, Prié D,
Courbebaisse M. Hyperuricémie chez les patients drépanocytaires suivis
en France. La Revue de Médecine Interne.2012 ; 33(1) :13-17. Doi :
10.1016/j.revmed.2011.07.002 https://doi.org/10.1016/j.revmed.2011.07.002 PMid:21907467
- Kaspar
CDW, Beach I, Newlin J, Sisler I, Feig D, Smith W. Hyperuricemia is
associated with a lower glomerular filtration rate in pediatric SCD
patients. Pediatr Nephrol. 2020;35(5):883-889.
doi:10.1007/s00467-019-04432-2 https://doi.org/10.1007/s00467-019-04432-2 PMid:31960140
- Gupta
S, Yui JC, Xu D, Fitzhugh CD, Clark C, Siddiqui S, Conrey AK, Kato GJ,
& Minniti C P. Gout and SCD: not all pain is sickle cell pain.
British Journal of Haematology, 171(5), 872-875. doi:10.1111/bjh.13433 https://doi.org/10.1111/bjh.13433 PMid:25892648 PMCid:PMC4699866
- Kenneth
I. Ataga, Vimal K. Derebail and David R. Archer. The glomerulopathy of
SCD: Am J Hematol.2014;89(9):907-914.doi:10.1002/ajh.23762 https://doi.org/10.1002/ajh.23762 PMid:24840607 PMCid:PMC4320776
- AIsaza-López
MC, Rojas-Rosas LF, Echavarría-Ospina L, Serna-Higuita LM.
Characterization of kidney complications in patients with sickle cell
anemia. Rev Chil Pediatr. 2020;91(1):51-57. Doi:
10.32641/rchped.v91i1.1274 https://doi.org/10.32641/rchped.v91i1.1274 PMid:32730413
- Ephraim
RK, Osakunor DN, Cudjoe O, Oduro EA, Asante-Asamani L, Mitchell J,
Agbodzakey H, Adoba P. Chronic kidney disease is common in SCD: a
cross-sectional study in the Tema Metropolis, Ghana. BMC Nephrol.
2015;16:75. doi:10.1186/s12882-015-0072-y https://doi.org/10.1186/s12882-015-0072-y PMid:26021375 PMCid:PMC4448314
- Aloni
MN, Ngiyulu RM, Gini-Ehungu JL, Nsibu CN, Ekila MB, Lepira FB, Nseka
NM. Renal function in children suffering from SCD: challenge of early
detection in highly resource-scarce settings. PloS one, 9(5),
e96561. https://doi.org/10.1371/journal.pone.0096561 PMid:24810610 PMCid:PMC4014510
- Thompson
J, Reid M, Hambleton I, Serjeant GR. Albuminuria and renal function in
homozygous SCD: observations from a cohort study. Arch Intern Med.
2007;167(7):701-708. doi:10.1001/archinte.167.7.701 https://doi.org/10.1001/archinte.167.7.701 PMid:17420429
- Ranque
B, Menet A, Diop IB, Thiam MM, Diallo D, Diop S, Diagne I, Sanogo I,
Kingue S, Chelo D, Wamba G, Diarra M, Anzouan JB, N'Guetta R, Diakite
CO, Traore Y, Legueun, G, Deme-Ly I, Belinga S, Boidy K, Jouven X.
Early renal damage in patients with SCD in sub-Saharan Africa: a
multinational, prospective, cross-sectional study. Lancet Haematol.
2014;1(2):e64-e73. doi:10.1016/S2352-3026(14)00007-6 https://doi.org/10.1016/S2352-3026(14)00007-6
- McPherson
Yee M, Jabbar SF, Osunkwo I, Clement L, Lane PA, Eckman JR, Guasch A.
Chronic kidney disease and albuminuria in children with SCD. Clin J Am
Soc Nephrol. 2011;6(11):2628-2633. doi:10.2215/CJN.01600211 https://doi.org/10.2215/CJN.01600211 PMid:21940843 PMCid:PMC3359567
- AlAmeer
MR, Alsarhan BK, Alsarhan LK, Albeshi SM, Alhenaki GA, Alqhtani MM,
Alasmari HR, Alabdali AH, Alsaleh TA, Alyami NM, Almansour AM, Almqaadi
AK, Alhazmi AA. Epidemiology of sickle cell nephropathy in sickle cell
anemia children, Saudi Arabia. Medical Science, 2021; 25(112),
1486-1493.
- Gosmanova EO, Zaidi S, Wan
JY, Adams-Graves PE. Prevalence and progression of chronic kidney
disease in adult patients with SCD. J Investig Med. 2014;62(5):804-07.
doi:10.1097/01.JIM.0000446836.75352.72 https://doi.org/10.1097/01.JIM.0000446836.75352.72 PMid:24781553
- Adebayo
OC, Van den Heuvel LP, Olowu WA, Levtchenko EN, Labarque V. Sickle cell
nephropathy: insights into the pediatric population [published online
ahead of print, 2021 May 29]. Pediatr Nephrol.
2021;10.1007/s00467-021-05126-4. doi:10.1007/s00467-021-05126-4 https://doi.org/10.1007/s00467-021-05126-4
- Laurin
LP, Nachman PH, Desai PC, Ataga KI, Derebail VK. Hydroxyurea is
associated with lower prevalence of albuminuria in adults with SCD.
Nephrol Dial Transplant. 2014;29(6):1211-1218. doi:10.1093/ndt/gft295 https://doi.org/10.1093/ndt/gft295 PMid:24084325 PMCid:PMC4038249
- Kim
NH, Pavkov ME, Knowler WC, Hanson RL, Weil EJ, Curtis JM, Bennett PH,
Nelson RG. Predictive value of albuminuria in American Indian youth
with or without type 2 diabetes. Pediatrics. 010;125(4):e844-e851.
doi:10.1542/peds.2009-1230 https://doi.org/10.1542/peds.2009-1230 PMid:20194283 PMCid:PMC3481836
[TOP]