Antonio Giordano1 and Livio. Pagano1,2.
1 Department
of Hematology, Fondazione Policlinico Universitario Agostino Gemelli –
IRCCS, Largo A. Gemelli, 8 I-00168 Rome, Italy.
2 Università Cattolica del Sacro Cuore, Largo A. Gemelli, 8 I-00168 Rome, Italy.
Published: March 1, 2022
Received: November 26, 2021
Accepted: February 16, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022029 DOI
10.4084/MJHID.2022.029
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Cutaneous
T-cell lymphomas are a heterogeneous group of T-cell neoplasms
involving the skin, the majority of which may be classified as Mycosis
Fungoides (MF) or Sézary Syndrome (SS).
Mycosis fungoides (MF) is
usually associated with an indolent clinical course and intermittent,
stable, or slow progression of the lesions. Extracutaneous involvement
(lymph nodes, blood, or less commonly other organs) or large cell
transformation (LCT) may be seen in advanced-stage disease. Sezary
syndrome (SS) is a rare leukemic subtype of CTCL characterized by
significant blood involvement, erythroderma, and often lymphadenopathy.
Although
the early-stage disease can be effectively treated predominantly with
skin-directed therapies, systemic therapy is often necessary to treat
advanced-stage disease. Systemic therapy options have evolved in recent
years with the approval of novel agents such as vorinostat, brentuximab
vedotin, and mogamulizumab. This review aims to discuss the diagnosis
and management of advanced-stages MF and SS.
|
Introduction
Mycosis
fungoides (MF) and Sezary syndrome (SS) are the most common variants of
cutaneous T-cell lymphoma (CTCL), which represent, in the Western
world, ∼75% to 80% of all primary cutaneous lymphomas, being the B
cutaneous lymphoma 20% to 25% The prognosis of MF and SS depends
on the type and extent of skin lesions and extracutaneous disease,
which were first captured in the TNM classification published for CTCL
in 1979. Suggested modifications published in 2007 for MF/SS revised
the nodal clinicopathologic classification adding blood involvement to
the staging of MF/SS.[1]
Mycosis fungoides (MF) is the most
common subtype and is usually associated with an indolent clinical
course with intermittent, stable, or slow progression of the lesions.
Extracutaneous involvement (lymph nodes, blood, or less commonly other
organs) or large cell transformation (LCT)[5] may be seen in
advanced-stage disease. Sézary syndrome (SS) is a rare erythrodermic,
leukemic variant characterized by significant blood involvement,
erythroderma, and often lymphadenopathy.[1]
The incidence of CTCL
has increased in recent decades; currently, it is 6.4 per 1,000,000
people with a median age of presentation 55-60 years old, predominantly
Caucasian males. Retrospective epidemiological studies have shown that
African-American, Hispanic, and Middle Eastern individuals may have a
higher incidence of CTCL (especially MF) than white individuals and
younger age and more aggressive course.[2]
MF is caused by the
malignant transformation of skin-resident effector memory T cells,
while SS is thought to arise from thymic memory T cells, supporting the
contention that SS is a process distinct from MF. However, cases
presenting as an overlap of these two conditions also exist.[3]
Folliculotropic
MF (FMF), granulomatous slack skin, and pagetoid reticulosis are
recognized as distinct clinicopathologic variants of MF in the
WHO-EORTC classification.[1]
SS is defined by a triad of
erythroderma, generalized lymphadenopathy, and the presence of clonally
related neoplastic T cells with cerebriform nuclei (Sezary cells) in
the skin, lymph nodes, and peripheral blood.[4] This review describes
systemic approaches for advanced-stage disease (stage IIB-IV).
Staging, Molecular Biology, and Prognosis
Adequate staging should be carried out to exclude the presence of extracutaneous disease.
Initial
work-up for patients with MF/SS also includes a complete physical
examination, representative skin biopsy, complete and differential
blood cell count, routine serum biochemistry with lactate dehydrogenase
(LDH), and appropriate imaging studies (CT and/or FDG-PET scans).[6]
Bone marrow biopsy and aspirate should be carried out in cutaneous
lymphomas with an intermediate or aggressive clinical behavior but is
not required in cutaneous lymphomas with an indolent clinical behavior
unless indicated by other staging assessments.[7]
Flow cytometry of the peripheral blood is usually recommended for all stages of MF.
The
following immunophenotypes characterize MF and SS cells: CD2+, CD3+,
CD5+, CD4+, CD8-, CCR4+, TCR-beta+, and CD45RO+ and absence of certain
T-cell markers, CD7 and CD26. However, there are subtypes of MF that
are CD8+ (especially the hypopigmented variant) or CD4/CD8
double-negative (in those with LCT), although rare.[7,8]
For
clinical staging of MF and SS, the revised tumor, node, metastasis, and
blood (TNMB) staging system should be used. Apart from the clinical
stage, older age, large cell transformation, and increased LDH values
have been identified as independent unfavorable prognostic factors in
MF.[8,9]
Despite some uncontrolled clinical trial results that
have been reported to suggest "cures" in this disease, the general
perception remains that this disease is not curable with standard
therapies available today. The disease behaves similarly to other
low-grade lymphomas, with periods of remission gradually becoming
shorter with subsequent therapeutic interventions. Patients with
significant nodal involvement (N3 or N4) or extensive skin involvement
(T4) have median life expectancies of 30-55 months.[10] Therefore, a
driving force in the development of treatments for this disease is
altering the natural history of this group of poor prognosis patients.
Recently, through the next generation sequencing (NGS), we have
understood the mutational profile that underlies the pathogenesis of
cutaneous T-cell lymphomas, and specifically, we have identified the
fundamental genetic and epigenetic alterations. Within pathogenetic
mechanisms, the role of T-cell clones with the presence of inflammatory
cytokines related to the TH2 profile is very important to favor both
the dysregulation of the immune system with a consequent deficit of
immunosurveillance and the creation of a favorable microenvironment.
Furthermore, there are numerous cytokines involved in addition to
Th2-secretion related, particularly IL-10, IL-15, IL-16, IL-17A,
IL-17F, IL-22, and IL-32 which have the primary purpose of suppressing
the immunological response regarding the tumor immunosurveillance
function. From the molecular point of view, the cellular stimulation
mediated by cytokines and chemokines generates TH2 based inflammatory
context with constitutive activation of the STAT pathway and loss of
complexity of the TCR. Therefore, forming a clonal population of T
cells with specific genetic-molecular alterations results in precarious
equilibrium with the cellular and humoral part of the
microenvironment.[11]
In 2007 staging system was revised by the
International Society for Cutaneous Lymphomas (ISCL) and the EORTC to
incorporate advances related to tumor cell biology and diagnostic
techniques, including the status of blood involvement. Investigators at
the National Cancer Institute retrospectively analyzed 152 patients who
underwent uniform pathologic staging. They were able to identify three
distinct prognostic groups. Good-risk patients had plaque-only skin
disease without lymph node, blood, or visceral involvement and a median
survival of more than 12 years. Less than 10% of patients with stage 1A
(localized patches) and less than 30% with stage 1B (extensive patch or
plaque) progress to more advanced disease. Intermediate-risk patients
had skin tumors, erythroderma, or plaque disease with lymph node or
blood involvement (but no visceral disease) and a median survival of 5
years. Poor-risk patients had a visceral disease or complete effacement
of lymph nodes by lymphoma, and a median survival of 2.5 years.[12]
Cytogenetic
analysis precisely identifies the individual chromosomal structure and
number. Bunn et al. demonstrated that in MF/SS, the presence of
aneuploidy karyotype during the clinical course was associated with
more aggressive disease. Hyperdiploid cell clones were demonstrated in
patients with large-cell histology, aggressive disease, and shortened
survival time.
Specific chromosomal deletions also influenced prognosis.[13]
Currently,
there are no valid markers to measure the prognosis of patients with
cutaneous T-cell lymphoma. However, in a recent paper, Shen et al.
showed that miR-155 and miR-200b expression in association with
elevated Ki-67 was significantly associated with worsening overall
survival in CTCL patients. Through this association, it was possible to
create a risk score classification projected on 5-year survival.[14]
Furthermore, the identification of this mechanism and the understanding
of epigenetic phenomena in the pathogenesis of LCT-MF have determined a
potential therapeutic role. Notably, a phase 1 study of MRG-106
(Cobomarsen), an inhibitor of miR-155, demonstrated efficacy in
patients with MF.[14]
From the prognostic point of view, Di
Raimondo et al. demonstrated the specific expression of twelve miRNAs
in MF patients undergoing clinical transformation to LCT-MF, thus
identifying the possibility of early progression markers.[15]
The
nuclear contour index has been used by several groups to separate
"benign" cutaneous lymphocytic disorders, such as Lymphomatoid
Papulosis and Pityriasis Lichenoides, from MF/SS.[16]
Trearment
Most
patients with early-stage MF (stage I or IIA) follow an indolent
course, and in particular, patients with stage IA MF have a similar
life expectancy as age, sex, and race-matched control populations. For
early-stage MF, the treatment concept is to control skin lesions mainly
by skin-directed therapies, such as topical therapies, phototherapies,
and radiotherapies, with the lowest possible side effects.
Unfortunately, early aggressive therapy does not appear to improve
survival when compared with skin-directed therapies.[17]
Systemic Therapy
Currently,
systemic chemotherapy is reserved for those patients with relapsed or
refractory disease after topical interventions or for those patients
with advanced nodal or visceral disease at presentation.
Bexarotene
is available and is EMA-approved for the treatment of the skin
manifestations of advanced stage (IIB–IVB) CTCL in adult patients
refractory to at least one systemic treatment.[18] In the US, Bexarotene
is FDA-approved as a second-line treatment for the early and late-stage
refractory disease (IB–IVB).[19] The recommended initial dose is 300
mg/m2/day, and this is taken as a single oral daily dose with a meal.
The dose is adjusted up or down according to clinical response and
toxicity to a maximum of 650 mg/m2/day. In the poor responders,
Bexarotene may also be safely combined with other anti-CTCL therapies,
including PUVA, ECP, methotrexate, and alpha-interferon to augment
responses.[20] It is 99% protein-bound and metabolized by cytochrome P450
3A4 (CYP3A4) to hydroxybexarotene and oxybexarotene and excreted in the
bile. Therefore, it is recommended that Bexarotene should be avoided in
patients with hepatic impairment. Other contraindications include a
history of pancreatitis, hypervitaminosis A and pregnancy.
Older
agents studied previously include alkylating agents such as
chlorambucil or cisplatin, the microtubule inhibitors such as
etoposide, vincristine, and vinblastine, or the antitumor antibiotics,
such as bleomycin and doxorubicin. In general, the responses are
modest, and the duration of response is typically less than six months.
McDonald and Bertino reported particularly good results with the
antimetabolite methotrexate administered intravenously followed by oral
citrovorum factor. Patients received 1-5 mg/kg of intravenous
methotrexate every five days. If a patient tolerated the lowest dose,
each subsequent dose was escalated. After five intravenous doses,
patients were switched to oral methotrexate (25-50 mg) with oral
citrovorum as weekly maintenance. All 11 patients achieved "good" or
better clearing (>60%) for a median duration of 24 months. Mucositis
and skin ulcerations were the most significant toxicity witnessed.
Myelosuppression was mild in general.[21]
Gemcitabine monotherapy is
an effective treatment option resulting in an ORR of 48% to 71% in
patients with heavily pretreated advanced-stage MF and SS. In a
retrospective observational study of 25 patients with advanced MF and
SS, after a long-term follow-up of 15 years, the estimated OS, PFS, and
DFS rates were 47%, 9%, and 40%, respectively.[22] Gemcitabine
monotherapy has also demonstrated front-line therapy activity in
untreated MF and SS patients.
Pegylated liposomal doxorubicin has
shown single-agent activity in patients with pretreated, advanced, or
refractory MF and SS. In an EORTC multicenter trial (phase II) of 49
patients with advanced MF (stage IIB, IVA, IVB), relapsed/refractory
after at least two prior systemic therapies, liposomal doxorubicin
resulted in an ORR of 41% (6% CR). The median time to progression was
seven months, and the median duration of response was 6 months.
Pegylated liposomal doxorubicin was well tolerated with no grade 3 or 4
hematologic toxicities; the most common grade 3 or 4 toxicities
included dermatologic toxicity other than hand and foot reaction (6%),
constitutional symptoms (4%), gastrointestinal toxicities (4%), and
infection (4%).[23]
In phase III randomized study (ALCANZA),[24,25]
brentuximab vedotin (anti-CD30 antibody-drug conjugate) attained
clinical outcomes superior to physicians' choice of methotrexate or
Bexarotene in patients with previously treated CD30-expressing MF. In
this study, 131 patients with previously treated CD30-expressing MF and
primary cutaneous anaplastic large cell lymphoma (PC-ALCL) (97 patients
with MF) were randomized to receive either brentuximab vedotin
or physician's choice (methotrexate or Bexarotene). At a
median follow-up of 23 months, the primary endpoint, ORR lasting for ≥4
months, was significantly higher for brentuximab vedotin compared to
methotrexate or bexarotene in the intent-to-treat population (56% [16%
CR] vs. 13% [2% CR]; P < .0001). In addition, peripheral neuropathy
was the most common adverse event reported in 67% of patients treated
with brentuximab vedotin.[26]
Vorinostat 400 mg daily orally was
tested in an open-label trial of 74 patients who had progressed on at
least two prior systemic therapies. The ORR (skin only) was 29.5%, with
1 CR and 18 PRs. Common adverse events included diarrhea (49%) and
fatigue (46%). Grade 3 events were less common but included fatigue
(5%), deep venous thromboses/pulmonary emboli (5%), and
thrombocytopenia (4%). Reports from the National Cancer Institute with
romidepsin have provided confirmatory results by using this class of
agents to treat patients with T-cell lymphomas, including some with
MF/SS. In several phase I and II trials, 50% of patients with MF/SS
appeared to have had a PR. Two additional clinical trials demonstrated
activity in Cutaneous T Cell Lymphoma(CTCL).[27] Vorinostat is
currently only approved in the United States.
In general,
toxicity to romidepsin and vorinostat has included alterations in the
cardiac conduction that could potentially predispose to arrhythmias,
and treatment of patients has required ongoing telemetry monitoring in
some trials. However, no evidence for acute or chronic impairment in
cardiac function has been noted. Vorinostat therapy led to drug-related
grade 1 electrocardiographic changes in five patients and grade 2 in
one patient. Therefore, using these agents in the outpatient setting
appears safe with a periodic assessment of cardiac rhythm and QTc
interval with an electrocardiogram base.[28] Unfortunately, romidepsin is
a substrate for the MDR protein (a P-glycoprotein) and upregulates the
expression of MDR1. Preliminary molecular analyses confirmed the
upregulation of MDR1. These data suggest that when resistance to this
agent develops, other chemotherapeutic drugs handled by MDR1 may be
rendered ineffective.[29]
IFN-α is an active agent for the treatment
of MF. Dosages and routes of administration have differed among
studies. Initially, high-dose IFN was used, with maximum doses of 36-50
million International Units (IU). Bunn et al. and Olsen et al.
independently demonstrated complete response rates of 10%-27% in
heavily pretreated patients. However, the duration of response was only
5.5 months. Later trials of untreated patients with doses of 3-18
million IU given subcutaneously daily have demonstrated an overall
response rate of 80%-92%.[30] From all these studies, it appears that a
reasonable and tolerable single-agent dose is 12 million IU/m2
administered subcutaneously daily. We recommend starting at 3 million
IU and gradually increasing as the patient tolerates the treatment.
Side
effects of all IFNs are dose-dependent. The most common adverse effects
are constitutional symptoms: fever, chills, myalgias, malaise, and
anorexia. Rarely, cytopenias, elevations of liver function test
results, renal dysfunction, cardiac dysfunction, or changes in mental
status (psychiatric syndromes).
Recently Mogamulizumab, a
humanized anti-CCR4 monoclonal antibody, was approved for the treatment
of relapsed or refractory MF and SS after at least one prior systemic
therapy. The approval was based on a phase III randomized, open-label,
multicenter trial (MAVORIC).[31] In this trial, 372 eligible patients
with relapsed or refractory MF and SS were randomized to either
mogamulizumab (n = 186) or vorinostat (n = 186). Mogamulizumab resulted
in significantly higher investigator-assessed ORR (28% vs. 5%; P <
.0001) and superior investigator-assessed median PFS (8 months vs. 3
months; P < .0001) compared with vorinostat, after a median
follow-up of 17 months. The ORR was higher in patients with SS than in
those with MF (37% vs. 21%). Among the 186 patients randomly assigned
to vorinostat, 136 patients (109 patients with disease progression and
27 patients after intolerable toxicity) crossed over to mogamulizumab.
The ORR was 31% for the 133 patients who crossed over from vorinostat
to mogamulizumab and subsequently received mogamulizumab.
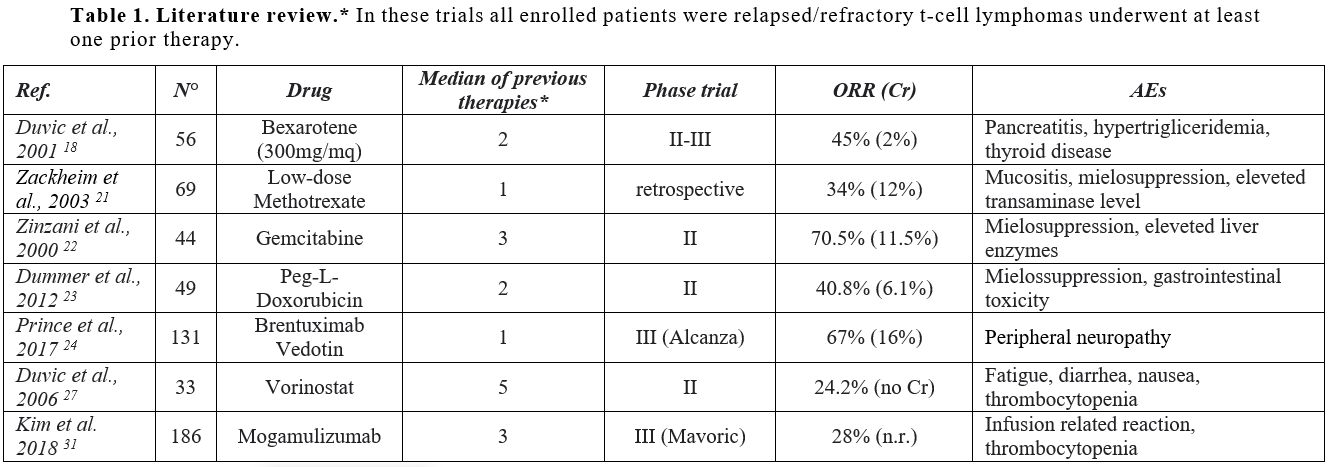 |
Table 1. Literature review.* In these trials all enrolled patients were relapsed/refractory t-cell lymphomas underwent at least one prior therapy. |
In the
post-hoc subgroup analysis by clinical stage, the ORRs for
mogamulizumab were higher for patients with stage III (23%) or stage IV
disease (36%) than those with stage IIB (16%) or stage IB/IIA disease
(19%). For skin, blood involvement, and lymph nodes, the
compartment-specific ORRs for mogamulizumab were 42%, 68%, and 17%,
respectively. The corresponding ORRs for vorinostat were 16%, 19%, and
4%, respectively. This trial, however, was not powered to detect OS
differences between the two groups within the defined follow-up
period.[32] The most common adverse events associated with mogamulizumab
were mostly graded 1-2 and manageable (infusion-related reactions
[37%], skin eruptions [25%], and diarrhea [14%]). Pyrexia (4%) and
cellulitis (3%) were the most common grade 3 adverse events in the
mogamulizumab group. Patients with the greatest symptom burden and
functional impairment took advantage, in terms of quality of life,
mostly from mogamulizumab.
In a phase II study of 24 patients with
MF and SS (stage IIB–IV) treated with at least one prior systemic
therapy, at a median follow-up of 40 weeks, pembrolizumab, an immune
checkpoint inhibitor, resulted in an ORR of 38% (the ORR was slightly
higher in patients with MF [56% vs. 27% for SS]) and a one-year PFS
rate of 65%. In addition, Pembrolizumab was associated with a skin
flare reaction, occurring exclusively in patients with SS. The flare
reaction correlated with high PD-1 expression on Sézary cells and
should be distinguished from disease progression.[33]
Role of Stem Cell Transplantation
Allogeneic
HCT has a role in a subset of patients with advanced-stage MF and SS
who have received multiple lines of therapy, as shown in retrospective
studies and small prospective series of patients with advanced MF and
SS.
In a multicenter retrospective analysis of 37 patients with
advanced-stage primary CTCL treated with allogeneic HCT (24 patients
[65%] had stage IV MFSS or disseminated nodal or visceral involvement),
after a median follow-up of 29 months, the incidence of relapse was
56%, and the estimated 2-year OS and PFS rates were 57% and 31%,
respectively.[34]
In a retrospective analysis of
patients with advanced-stage MF and SS in the European Group for Blood
and Marrow Transplantation (EBMT) database (n = 60) treated with
allogeneic HSCT, the 5-year PFS and OS rates were 32% and 46%,
respectively. The corresponding 7-year survival rates were 44% and 30%,
respectively. The non-relapse mortality (NRM) rate at 7 years was 22%.
Outcomes were not significantly different between histology types.
However, patients with advanced-stage disease had an increased risk of
relapse or progression and lower PFS, and myeloablative conditioning
was associated with poorer NRM and OS.
Besides, transplants from
unrelated donors had a statistically borderline impact on NRM and a
significantly lower PFS and OS. In a case series of 47 patients with
advanced-stage MF and SS who underwent allogeneic HCT after the failure
of standard therapy, the estimated 4-year OS and PFS rates were 51% and
26%, respectively. While there was no statistical difference in the OS
in patients who had MF without LCT, SS, MF with LCT, or SS with LCT,
the 4-year PFS rate was superior in patients who had SS versus those
who did not (52% vs.10%; P =
.02). Recent systematic reviews and meta-analyses have reported pooled
PFS and OS rates of 36% and 59%, respectively. Autologous HCT is not
recommended for patients with CTCL due to the short duration of
response despite its toxicity, thus limiting its utility.[35]
Emerging Therapies and Conclusion
The
advanced stages of mycosis fungoides still have a poor prognosis.
Current treatment options have improved the management of skin
manifestations without significantly increasing survival. In our
experience, conventional chemotherapy still plays a valid role,
especially in a high burden disease. The new therapies represented by
monoclonal antibodies, sometimes conjugated with cytotoxic agents, aim
to maximize the therapeutic effect through a biological target and
reduce adverse events. Notably, targeted therapy has shown some
interesting recent developments in many cancers and could have major
implications for CTCL.
Anti-drug conjugates, which target surface
markers such as CD30, have shown better results, although with a
toxicity profile that makes them unsuitable for all patient categories.
AFM13 is a chimeric antibody with an anti-CD30 murine domain. An
open-label Phase II multicenter study is underway to evaluate the
efficacy and safety of AFM13 in patients with transformed mycosis
fungoides (REDIRECT).
In addition, immune checkpoint inhibitors
such as anti-PD-1 should be considered in the treatment of CTCL.
Activation of innate immune mechanisms that support Th1 responses must
be investigated alone or in combination with depletion of malignant T
cells.
Finally, Zanolimumab is a humanized anti-CD4 + mAb
expressed on most T lymphocytes and is therefore useful in most CD4+
lymphoproliferative diseases. Kim et al. described 47
relapsed/refractory MF/SS patients in two phase II trials that
showed a high response rate in the maximum dose group (56%) with a
median duration of response of 81 weeks.[36]
Therefore,
we needed further studies to understand the targeted therapy's timing
and possibly combination treatments. Nevertheless, the use of the
molecular target is certainly a valid strategy to reduce the minimum
measurable disease and confer an advantage on consolidation treatments,
especially concerning
References
- Willemze R, Cerroni L, Kempf W, et al. The 2018
update of the WHO-EORTC classification for primary cutaneous lymphomas.
Blood 2019;133:1703-1714. https://doi.org/10.1182/blood-2018-11-881268 PMid:30635287 PMCid:PMC6473500
- Wilson
LD, Hinds GA, Yu JB. Age, race, sex, stage, and incidence of cutaneous
lymphoma. Clin Lymphoma Myeloma Leuk. 2012 Oct;12(5):291-6. doi:
10.1016/j.clml.2012.06.010. https://doi.org/10.1016/j.clml.2012.06.010 PMid:23040434 PMCid:PMC3475508
- Campbell
JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis
fungoides arise from distinct T-cell subsets: a biologic rationale for
their distinct clinical behaviors. Blood 2010;116:767-771. https://doi.org/10.1182/blood-2009-11-251926 PMid:20484084 PMCid:PMC2918332
- Olsen
EA, Rook AH, Zic J, et al. Sezary syndrome: immunopathogenesis,
literature review of therapeutic options, and recommendations for
therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J
Am Acad Dermatol 2011;64:352-404. https://doi.org/10.1016/j.jaad.2010.08.037 PMid:21145619
- Vergier
B, de Muret A, Beylot-Barry M, et al. Transformation of mycosis
fungoides: clinicopathological and prognostic features of 45 cases.
French Study Group of Cutaneous Lymphomas. Blood 2000;95:2212-2218.
- Olek-Hrab K, Silny W. Diagnostics in mycosis fungoides and Sezary syndrome. Rep Pract Oncol Radiother. 2013;19(2):72-76. https://doi.org/10.1016/j.rpor.2013.11.001 PMid:24936324 PMCid:PMC4054990
- Olsen
E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and
classification of mycosis fungoides and Sezary syndrome: a proposal of
the International Society for Cutaneous Lymphomas (ISCL) and the
cutaneous lymphoma task force of the European Organization of Research
and Treatment of Cancer (EORTC). Blood 2007;110:1713-1722. https://doi.org/10.1182/blood-2007-03-055749 PMid:17540844
- Agar
NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic
factors in mycosis fungoides/Sezary syndrome: validation of the revised
International Society for Cutaneous Lymphomas/European Organisation for
Research and Treatment of Cancer staging proposal. J Clin Oncol
2010;28:4730-4739. https://doi.org/10.1200/JCO.2009.27.7665 PMid:20855822
- Scarisbrick
JJ, Prince HM, Vermeer MH, et al. Cutaneous Lymphoma International
Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and
Sezary Syndrome: Effect of Specific Prognostic Markers on Survival and
Development of a Prognostic Model. J Clin Oncol 2015;33:3766-3773. https://doi.org/10.1200/JCO.2015.61.7142 PMid:26438120 PMCid:PMC4979132
- LeBlanc
RE, Lefterova MI, Suarez CJ et al. Lymph node involvement by mycosis
fungoides and Sézary syndrome mimicking angioimmunoblastic T-cell
lymphoma. Hum Pathol. 2015 Sep;46(9):1382-9. https://doi.org/10.1016/j.humpath.2015.05.024 PMid:26193796
- Patil
K, Kuttikrishnan S, Khan AQ, et al. Molecular pathogenesis of Cutaneous
T cell Lymphoma: Role of chemokines, cytokines, and dysregulated
signaling pathways. Semin Cancer Biol. 2021 Dec
11:S1044-579X(21)00296-0. doi: 10.1016/j.semcancer.2021.12.003. https://doi.org/10.1016/j.semcancer.2021.12.003
- Olsen
E, Vonderheid E, Pimpinelli Net al. ISCL/EORTC. Revisions to the
staging and classification of mycosis fungoides and Sezary syndrome: a
proposal of the International Society for Cutaneous Lymphomas (ISCL)
and the cutaneous lymphoma task force of the European Organization of
Research and Treatment of Cancer (EORTC). Blood. 2007 Sep
15;110(6):1713-22. doi: 10.1182/blood-2007-03-055749. Epub 2007 May 31.
Erratum in: Blood. 2008 May 1;111(9):4830. https://doi.org/10.1182/blood-2008-02-142653 PMID: 17540844
- Bunn
PA Jr, Whang-Peng J, Carney DN, Schlam ML, Knutsen T, Gazdar AF. DNA
content analysis by flow cytometry and cytogenetic analysis in mycosis
fungoides and Sézary syndrome. J Clin Invest. 1980 Jun;65(6):1440-8.
doi: 10.1172/JCI109808. https://doi.org/10.1172/JCI109808 PMid:6997334 PMCid:PMC371482
- Shen
X, Wang B, Li K, et al. MicroRNA Signatures in Diagnosis and Prognosis
of Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2018
Sep;138(9):2024-2032. doi: 10.1016/j.jid.2018.03.1500. Epub 2018 Mar
17. https://doi.org/10.1016/j.jid.2018.03.1500 PMid:29559342
- Di
Raimondo C, Han Z, Su C, et al. Identification of a Distinct miRNA
Regulatory Network in the Tumor Microenvironment of Transformed Mycosis
Fungoides. Cancers (Basel). 2021 Nov 22;13(22):5854. doi:
10.3390/cancers13225854. https://doi.org/10.3390/cancers13225854 PMid:34831008 PMCid:PMC8616450
- Shum
DT, Roberts JT, Smout MS, Wells GA, Simon GT. The value of nuclear
contour index in the diagnosis of mycosis fungoides. An assessment of
current ultrastructural morphometric diagnostic criteria. Cancer. 1986
Jan 15;57(2):298-304. doi:
10.1002/1097-0142(19860115)57:2<298::aid-cncr2820570218>3.0.co;2-1.
https://doi.org/10.1002/1097-0142(19860115)57:2<298::AID-CNCR2820570218>3.0.CO;2-1 PMID: 3942961.
- Kaye
FJ, Bunn PA, Steinberg SM, et al. A randomized trial comparing
combination electron-beam radiation and chemotherapy with topical
therapy in the initial treatment of mycosis fungoides. N Engl JMed.
1989;321(26):1784-90. https://doi.org/10.1056/NEJM198912283212603 PMid:2594037
- Duvic
M, Hymes K, Heald P et al. Bexarotene is effective and safe for
treatment of refractory advanced-stage cutaneous T-cell lymphoma:
multinational phase II-III trial results. J Clin Oncol 2001; 19:
2456-2471. https://doi.org/10.1200/JCO.2001.19.9.2456 PMid:11331325
- Duvic
M, Martin AG, Kim Y et al. Phase 2 and 3 clinical trial of oral
Bexarotene (Targretin capsule s) for the treatment of refractory or
persistent early-stage cutaneous T-cell lymphoma. Arch Dermatol 2001;
137: 581-593.
- Whittaker S, Oritz P,
Dummer R et al. Efficacy and safety of Bexarotene combined with
psoralen-ultraviolet A (PUVA) compared with PUVA treatment alone in
stage IB-IIA mycosis fungoides: final results from the EORTC Cutaneous
Lymphoma Task Force phase III randomized clinical trial (NCT00056056).
Br J Dermatol 2012; 163: 678-687. https://doi.org/10.1111/j.1365-2133.2012.11156.x PMid:22924950
- Zackheim
HS, Kashani-Sabet M, McMillan A. Low-dose methotrexate to treat mycosis
fungoides: a retrospective study in 69 patients. J Am Acad Dermatol
2003;49:873-878. https://doi.org/10.1016/S0190-9622(03)01591-3
- Zinzani
PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated
cutaneous T-cell lymphoma: experience in 44 patients. J Clin Oncol
2000;18:2603-2606. https://doi.org/10.1200/JCO.2000.18.13.2603 PMid:10893292
- Dummer
R, Quaglino P, Becker JC, et al. Prospective international multicenter
phase II trial of intravenous pegylated liposomal doxorubicin
monochemotherapy in patients with stage IIB, IVA, or IVB advanced
mycosis fungoides: final results from EORTC 21012. J Clin Oncol
2012;30:4091-4097. https://doi.org/10.1200/JCO.2011.39.8065 PMid:23045580
- Prince
HM, Kim YH, Horwitz SM, et al. Brentuximab vedotin or physician's
choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an
international, open-label, randomized, phase 3, multicentre trial.
Lancet 2017;390:555-566. https://doi.org/10.1016/S0140-6736(17)31266-7
- Duvic
M, Tetzlaff MT, Gangar P, et al. Results of a Phase II Trial of
Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and
Lymphomatoid Papulosis. J Clin Oncol 2015;33:3759-3765. https://doi.org/10.1200/JCO.2014.60.3787 PMid:26261247 PMCid:PMC4737859
- Dummer
R, Prince HM, Whittaker S, et al. Patient-reported quality of life in
patients with relapsed/refractory cutaneous T-cell lymphoma: Results
from the randomized phase III ALCANZA study. Eur J Cancer
2020;133:120-130. https://doi.org/10.1016/j.ejca.2020.04.010 PMid:32502876
- Duvic
M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat
(suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell
lymphoma (CTCL). Blood 2007;109:31-39. https://doi.org/10.1182/blood-2006-06-025999 PMid:16960145 PMCid:PMC1785068
- Duvic
M, Olsen EA, Breneman D, et al. Evaluation of the long-term
tolerability and clinical benefit of vorinostat in patients with
advanced cutaneous T-cell lymphoma. Clin Lymphoma yeloma
2009;9:412-416. https://doi.org/10.3816/CLM.2009.n.082 PMid:19951879
- Piekarz
RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the
histone deacetylase inhibitor romidepsin as monotherapy for patients
with cutaneous T-cell lymphoma. J Clin Oncol 2009;27:5410-5417. https://doi.org/10.1200/JCO.2008.21.6150 PMid:19826128 PMCid:PMC2773225
- Olsen EA. Interferon in the treatment of cutaneous T-cell lymphoma. Dermatol Ther 2003;16:311-321. https://doi.org/10.1111/j.1396-0296.2003.01643.x PMid:14686974
- Kim
YH, Bagot M, Pinter-Brown L, et al. Mogamulizumab versus vorinostat in
previously treated cutaneous T-cell lymphoma (MAVORIC): an
international, open-label, randomized, controlled phase 3 trial. Lancet
Oncol 2018;19:1192-1204. https://doi.org/10.1016/S1470-2045(18)30379-6
- Cowan
RA, Scarisbrick JJ, Zinzani PL et al. Efficacy and safety of
mogamulizumab by patient baseline blood tumour burden: a post hoc
analysis of the MAVORIC trial. J Eur Acad Dermatol Venereol. 2021
Nov;35(11):2225-2238. doi: 10.1111/jdv.17523. https://doi.org/10.1111/jdv.17523 PMid:34273208
- Khodadoust
MS, Rook AH, Porcu P, et al. Pembrolizumab in Relapsed and Refractory
Mycosis Fungoides and Sezary Syndrome: A Multicenter Phase II Study. J
Clin Oncol 2020;38:20-28. https://doi.org/10.1200/JCO.19.01056 PMid:31532724 PMCid:PMC6943974
- Duarte
RF, Boumendil A, Onida F, et al. Long-term outcome of allogeneic
hematopoietic cell transplantation for patients with mycosis fungoides
and Sezary syndrome: a European society for blood and marrow
transplantation lymphoma working party extended analysis. J Clin Oncol
2014;32:3347-3348. https://doi.org/10.1200/JCO.2014.57.5597 PMid:25154828
- Lechowicz
MJ, Lazarus HM, Carreras J, et al. Allogeneic hematopoietic cell
transplantation for mycosis fungoides and Sezary syndrome. Bone Marrow
Transplant 2014;49:1360-1365. https://doi.org/10.1038/bmt.2014.161 PMid:25068422 PMCid:PMC4221526
- Youn
H. Kim, Duvic M, Obitz E, et al. Clinical efficacy of Zanolimumab
(HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell
lymphoma, Blood, Volume 109, Issue 11, 2007, Pages 4655-4662,ISSN
0006-4971, https://doi.org/10.1182/blood-2006-12-062877 PMid:17311990
[TOP]