We describe a cross-sectional study involving 14 patients (7 with moyamoya syndrome and 7 with moyamoya disease), female: male 1.33:1, with a median age of 9.3 years (1.7-12.6) at diagnosis. Their median follow-up time was 6.3 years. The diagnosis was confirmed using magnetic resonance imaging and angiographic study. Recurrent hemiparesis and seizure were the most common presentations. All patients had no family history of moyamoya. Genomic DNA was extracted from peripheral blood samples and sent for WES, performed on a Novaseq 6000 System (Illumina, San Diego, CA, USA). WES data were analyzed using the commercial software, Sophia DDM, V4.4 (Sophia Genetics). This study was approved by the Ethics Committee of the Faculty of Medicine Ramathibodi Hospital (ID: COA. MURA2020/1788).
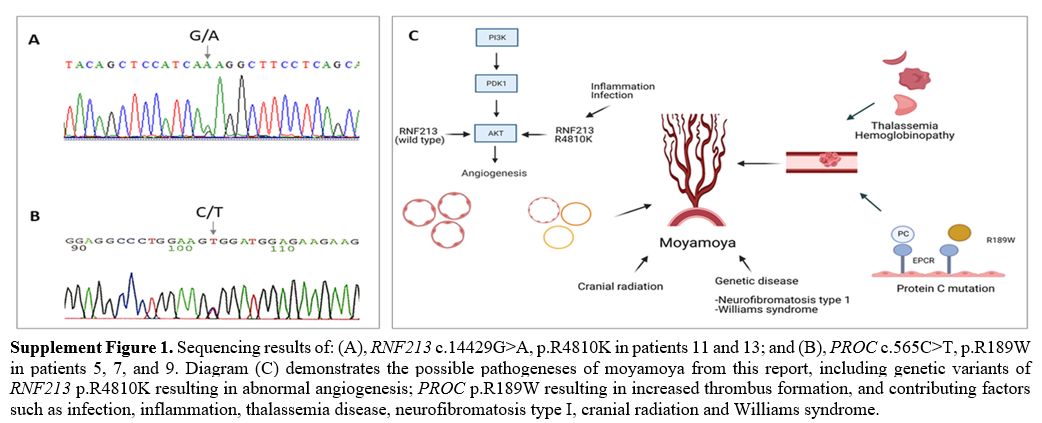
Among the seven patients with moyamoya syndrome, three were diagnosed with non-deletional α-thalassemia disease [hemoglobin H/Constant Spring (--SEA/CS)], one with Williams syndrome, and one post cranial radiation. Two of the three α-thalassemia patients also had APAs. The WES study demonstrated a heterozygous variant of a prothrombotic gene of heterozygous PROC gene mutation (c.565C > T, p.R189W) in three patients. Two patients had low protein (PC) activities (42% and 61%). WES study also confirmed the genetic information of thalassemia [αCS (HBA2: c.427T > C)/ --SEA (NG_000006.1: g.26264_45564del19301)] among three patients. One of the three heterozygous p.R189W mutation patients also had homozygous HBB c.79G>A, p.E27K, causing mild anemia (Table 1 and Supplement Figure 1). For the seven patients with moyamoya disease, the WES study identified genetic variants of heterozygous RNF213 mutation (c.14429G>A, p.R4810K) in two patients (Table 1 and Supplement Figure 1).
 |
Table 1. Characteristics of the 14 patients with moyamoya. Patients 1-7 were classified as moyamoya syndrome, and Patients 8-14 as moyamoya disease. |
 |
Neurological outcomes revealed neurologic deficits among seven patients. The modified Rankin scale (mRS) was higher in moyamoya syndrome (score 3 in craniopharyngioma post cranial radiation, score 2 in alpha thalassemia disease, and score 1 in Williams syndrome), indicating higher disability when compared with moyamoya disease (score 1 in two of seven patients).
Sickle cell anemia and β thalassemia had been reported in moyamoya syndrome.[1] The present study reported non-deletional α-thalassemia in moyamoya. The α-thalassemia disease usually involves moderate anemia and requires occasional red blood cell (RBC) transfusion. However, non-deletional α-thalassemia may exhibit more severe phenotypes, similar to patients with ꞵ-thalassemia.[3] The patients in this report, before the diagnosis of moyamoya, received occasional RBC transfusions. As a result, the high proportion of phosphatidylserine exposing RBC increased the hypercoagulable state[4] and may contribute to the developing moyamoya. The additional prothrombotic risk factors were APAs in two α-thalassemia patients and PROC p.R189W in one patient. The p.R189W mutation, resulted in low or slightly low levels of PC activity. The related report suggested that p.R189W had a low binding affinity to endothelial PC receptors; however, some patients with p.R189W had normal PC activity.[5,6] WES demonstrated the prothrombotic genetic variants of the PROC p.R189W mutation in three patients. These findings indicated the overall prothrombotic risk factors in 35.7% of patients and 71.4% of moyamoya syndrome, including two patients with APS and three with PROC p.R189W mutation. A related study reported prothrombotic risk factors consisting of APAs and PS deficiencies in 40% of the investigated ten patients.[7]
In addition to prothrombotic risk factors, one patient in this study presented Williams syndrome associated with vascular abnormality, including peripheral pulmonary stenosis and moyamoya syndrome.[1] One patient developed moyamoya syndrome after 18 months of 54 Gy cranial radiation. Altogether, 14.2% of our reported patients and 28.6% of moyamoya disease demonstrated RNF213 p.R4810K, which was lower than that reported among Japanese (90.1%) and Korean (78.9%). Still, the same as in Chinese (23.2%) patients.[2] The incidence was higher than the prevalence in the general population (0-11.4%).[2] The RNF213 gene, located on chromosome 17q25.3, is related to angiogenesis and vascular inflammation with an unknown physiologic function.
Although the number of enrolled patients was small due to the rarity of the disease, our report demonstrated a non-deletional type α-thalassemia disease (--SEA/ α^csα), and APAs in moyamoya syndrome. In addition to the prothrombotic risk factor of genetic variants of PROC p.R189W. Moreover, the (RNF213 p.R4810K was identified in 28.6% of moyamoya disease. In addition, the RNF213 p.R4810K was identified in 28.6% of moyamoya disease.