According to the International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS); ring sideroblasts (RS) are defined as erythroblasts in which there is a minimum of five siderotic granules covering at least one-third of the circumference of the nucleus.[3] SA is a rare disease affecting fewer than 200,000 people in the US [4]. Due to the low incidence and prevalence and the heterogeneity of the causative factors, no certain statistical data on the epidemiology of this disorder.
The SA is traditionally classified into congenital sideroblastic anemia (CSA) and acquired forms (ASA). ASA is further subclassified into two groups, clonal neoplastic disorders and benign disorders due to reversible metabolic factors.
Acquired clonal SA comprises myeloid stem cell disorders associated with RS, which was classified according to the 2016 revised World Health Organization (WHO) Classification into three categories: Myelodysplastic syndrome (MDS) with ring sideroblasts and single-lineage dysplasia (MDS-RS-SLD), MDS with ring sideroblasts and multilineage dysplasia (MDS-RS-MLD), and MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T). As their names suggest, the classification is based on detecting RS accompanied by dysplasia in one or multiple hematopoietic lineages, or anemia, thrombocytosis, and features of a myeloproliferative neoplasm.[2]
Conferring to WHO criteria: MDS-RS-SLD, MDS-RS-MLD, and MDS/MPN-RS-T ring sideroblasts should be >15% of the BM erythroid precursors; however, if SF3B1 mutation is detected, the diagnosis can be made with >5% ring sideroblasts marrow erythroid precursors.[2] For other listed categories of myeloid neoplasms, no specific cut-off RS percentage is required.
Mutations in the spliceosome that mediates the maturation of primary mRNA transcripts to mature mRNAs lacking introns have been identified as common in MDS-RS. Specifically, the acquired heterozygous somatic mutations in SF3b1 are the strongest molecular correlate of MDS-RS. SF3B1 mutations are present in between 70% and 90% of MDS-RS and, in many, it is the single detectable clonal marker.[5]
Non-neoplastic causes of RS include copper deficiency (which may be induced by a high dose of zinc administration), alcohol, toxins, and drugs (e.g., isoniazid).[6] Unlike in MDS-RS, patients with CSA tend to present at a much younger age and with microcytic (rather than macrocytic) anaemia.[7]
The design of this study is mixed (retrospective and prospective); we have analysed the clinical, pathologic, and molecular data of 15 cases of ASA diagnosed in Hamad Medical Corporate, in Qatar, between March 2015 and March 2022. The next generation sequence (NGS) panel designed to study targeted regions in 30 genes recurrently mutated in myeloid neoplasia (including SF3B1 mutations and TP53) was then performed (according to specimens' availability) and consequently analysed.
The study had ethical approval from IRB (Institutional research board; # MRC-04-22-209). Patients were waived from consent as it is a retrospective study.
ASA diagnosis was established by the presence of RS detected by Prussian blue staining (Perls' reaction) on BM aspirate smears (Figure 1). All included cases were examined independently by two experienced hematopathologists. Diagnosis and subclassification of ASA were made on BM specimens according to WHO 2016 classification. Relevant clinical, hematologic, and BM pathologic findings, including assessment of the percentage of trilineage dysplasia (a cut-off of 10%), flow cytometry immunophenotyping for myeloid neoplasms and cytogenetics data, were analysed. NGS panel for myeloid neoplasia was performed (according to specimens' availability) and consequently analysed. Clinical and laboratory data, including the disease course and outcome, were also retrieved and analyzed.
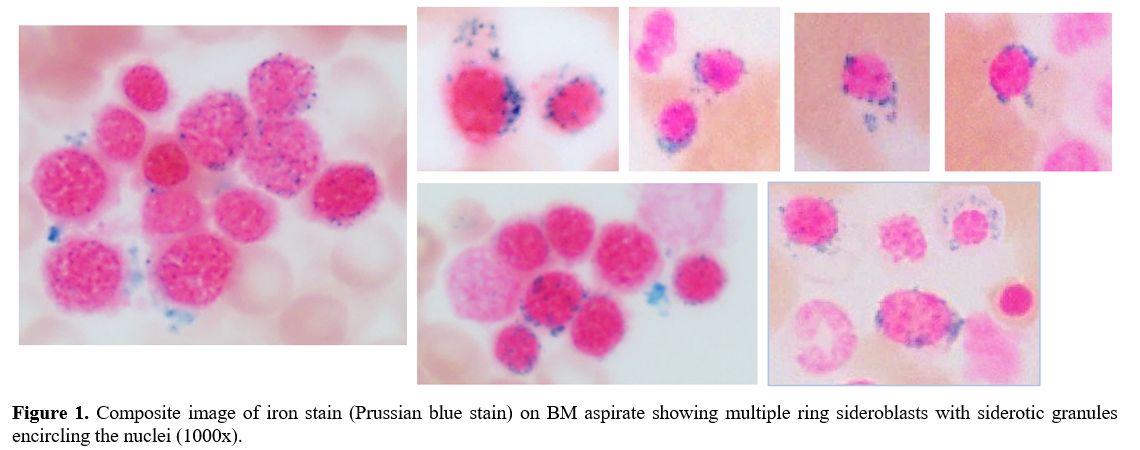 |
Figure 1. Composite image of iron stain (Prussian blue stain) on BM aspirate showing multiple ring sideroblasts with siderotic granules encircling the nuclei (1000x). |
Associations between two or more qualitative variables across two independent groups (clonal and non-clonal SA) were assessed using the Fisher Exact Chi-square test or Pearson Chi-square as appropriate. Quantitative data and outcomes measured across two groups were analysed using the Mann-Whitney U test (due to skewed or non-normal data distribution). All P values presented were two-tailed, and P values <0.05 were considered statistically significant. All Statistical analyses were performed using statistical packages SPSS version 27.0 (Armonk, NY: IBM Corp) and Epi-info (Center for Disease Control and Prevention, Atlanta, GA) software.
Fifteen patients of ASA were detected and clustered into two main groups: clonal SA (associated with a hematologic neoplasm) (10 cases, 66.7%) and SA secondary to non-clonal causes (5 cases, 33.3%). The latter included: SA Secondary to copper deficiency (two cases), SA secondary to pyridoxine deficiency, and the last group included two patients where the exact cause of SA remained unrevealed and classified accordingly as idiopathic SA (Tables 1 and 2).
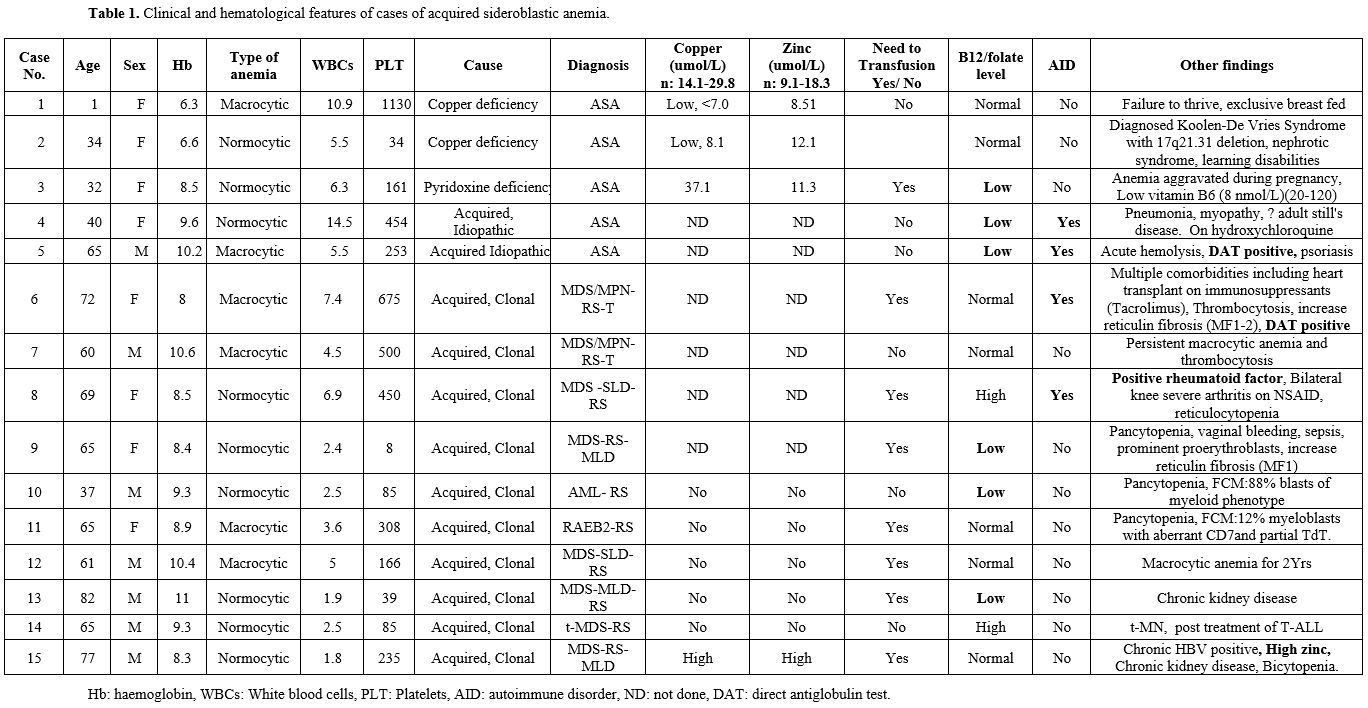 |
Table 1. Clinical and hematological features of cases of acquired sideroblastic anemia. |
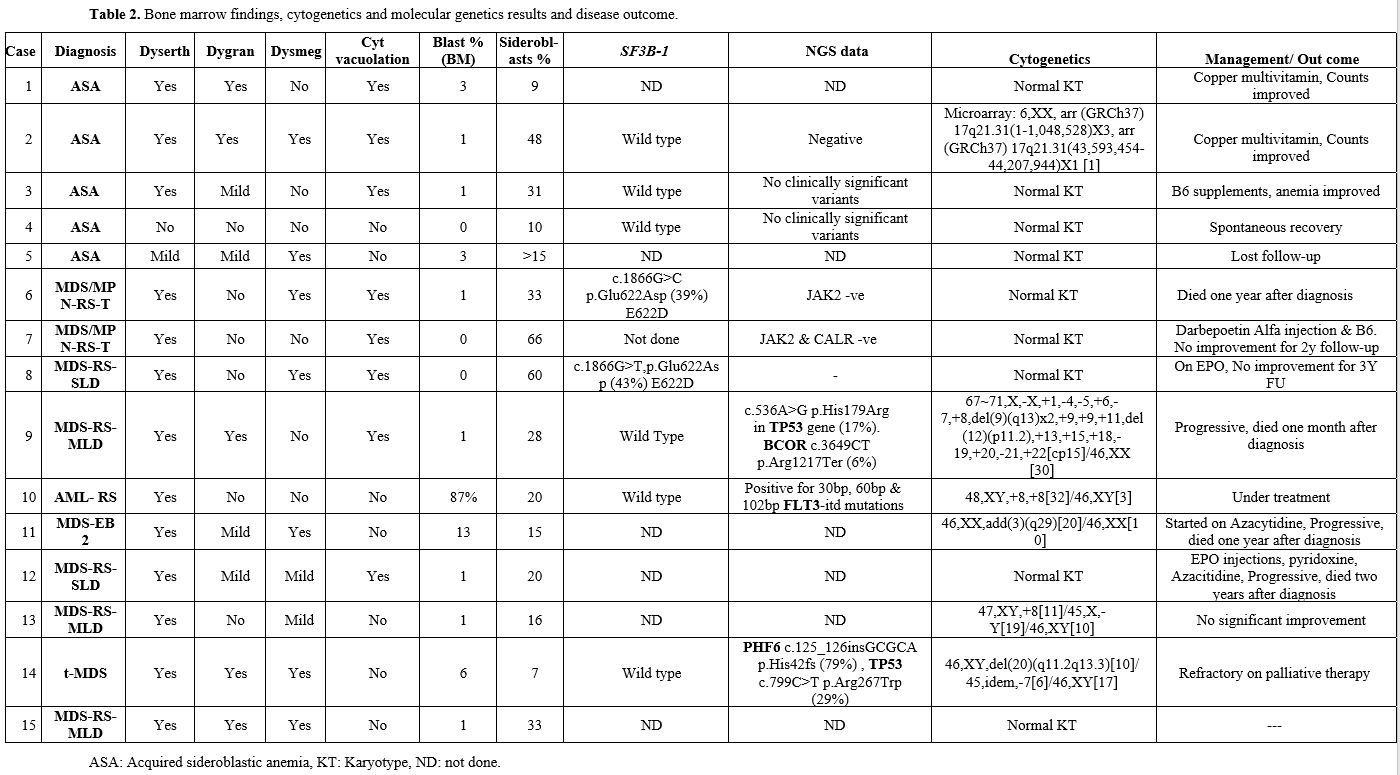 |
Table 2. Bone marrow findings, cytogenetics and molecular genetics results and disease outcome. |
ASA in our cohort was diagnosed in all age groups, with a median age of 65 years (IQR 37- 69 years), and almost equally represented between male and female gender. All patients had a significant degree of anemia; the median hemoglobin was 8.5 g/dL (IQR 8-10.2), 6 cases were macrocytic, and 8 were normocytic normochromic anemia. None of the cases in our cohort showed microcytic hypochromic anemia. The median white blood cell count (WBC) was 5×109 /L (IQR 2.5-6.9), and the median platelets count was 235 ×109 /L (IQR 39.0- 454).
Dyserythropoiesis was the most striking finding (in 14/15 (93.3%), mostly represented by cytoplasmic abnormalities: cytoplasmic irregularities/inclusions with prominent cytoplasmic vacuolation in 9/15 cases (60%). Dysmegakaryopoiesis was found in 9/15 patients (60%). Dysgranulopoiesis was detected in 8/15 (53.3%).
RS were detected at variable percentages ranging between 7-66% (median 20, IQR 13-33). Abnormal karyotype (KT) was detected in 5/14 (35.7%) myeloid neoplasms with RS. No cytogenetic abnormality was detected within the group of non-clonal SA.
NGS was performed in 8 out of 15 cases, including 3/5 of the non-clonal cases. SF3B1 mutatin was found in 2/8 patines (25%), one with MDS-SLD-RS and the other with MDS/MPN-RS-T. In six cases, SF3B1 was wild type. Further genetic mutations detected by NGS included the TP53 gene (case 9 & case 14), PHF6, FLT3-itd, and BCOR mutations.
Clinically, 60% (9/15) of ASA in our cohort had significant anemia that required regular blood transfusions. In addition, 6/15 (40%) cases had a concurrent low serum B12/folate. Associated autoimmune disorders, including DAT-positive autoimmune hemolytic anemia (two cases), adult Still's disease, and autoimmune arthritis, were observed in 4/15 patients (26.6%).
Comparison between acquired clonal (neoplastic) cases and non-clonal SA (Table 3). SA secondary to an associated myeloid neoplasm was predominant among the elderly, with 8 out of 9 cases diagnosed at an age > 60. The median age was 65 (IQR 60.75- 73.25 years), while non-clonal SA was more represented within the younger age group, with a median age of 34 (16.5-52.5 years). This difference was statistically significant (P=0.016). Interestingly, most non-clonal SA (80%) were female patients.
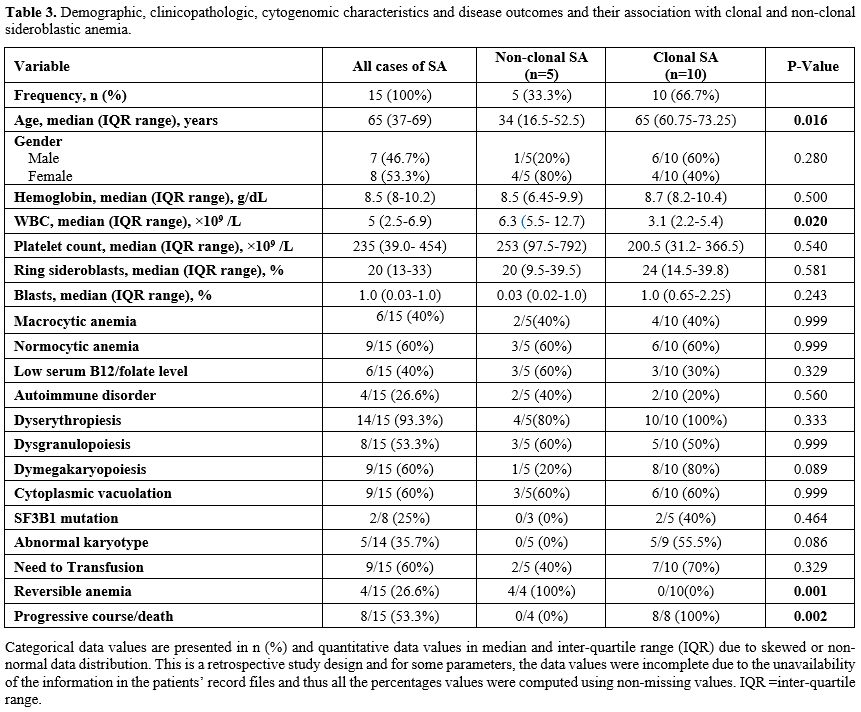 |
Table 3 Demographic, clinicopathologic, cytogenomic characteristics and disease outcomes and their association with clonal and non-clonal sideroblastic anemia. |
No statistically significant difference (P>0.05) was observed between the two groups regarding the haemoglobin, platelets count, or the type of anemia. However, WBC was significantly higher in non-clonal SA (median 6.3, IQR 5.5-12.7) compared to the clonal SA group (median 3.1, IQR 2.2-5.4), P=0.020.
Dyserythropoiesis and dysgranulopoiesis were equally present, with no significant morphologic difference between both groups regarding the type of dyserythropoietic changes. However, the cytoplasmic vacuolations were well defined and numerous in non-clonal SA (Figure 2A), while in clonal SA, there was a focal clearing of the cytoplasm rather than definitive vacuoles (Figure 3B). It is worth noting that the granulocytes in the group of non-clonal SA (especially those secondary to copper or pyridoxine deficiency) showed significant nuclear holes/well-defined defect/vacuolation within the nuclear chromatin (Figure 2B), in addition to the cytoplasmic vacuolations which were previously described in cases of copper deficiency. The latter finding was not appreciated within the MDS-associated SA, which showed abnormal granulation &/abnormal segmentation (Figure 3A).
Dysmegakaryopoiesis (Figure 3C) was most frequently detected among cases of clonal SA (8/10, 80%) compared to non-clonal cases (1/5, 20%). However, this difference was statistically insignificant (P=0.089). On the other hand, RS percentage tends to be higher in clonal SA, with no statistically significant difference (P=0.581).
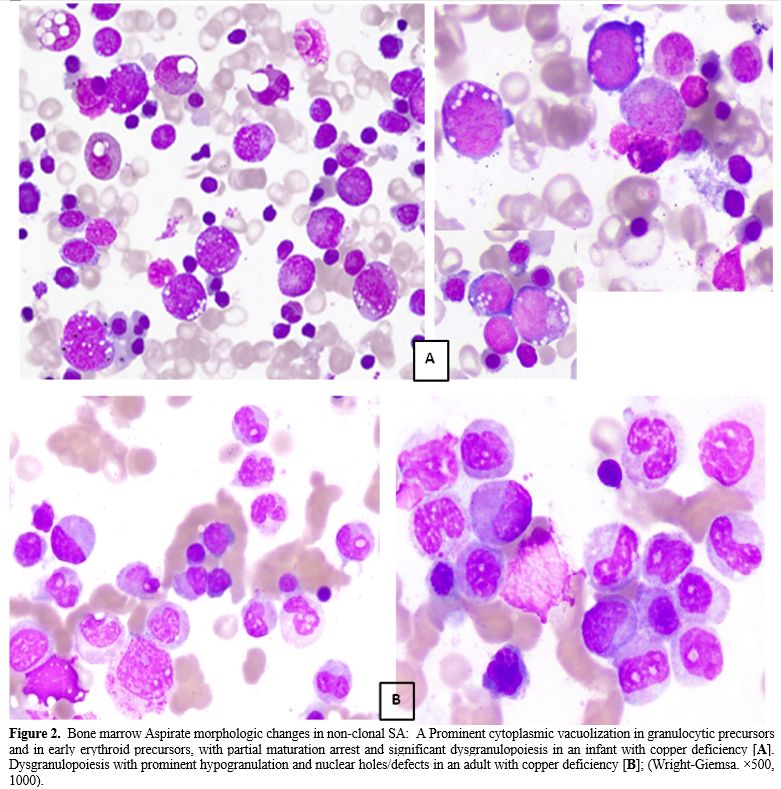 |
Figure 2. Bone marrow Aspirate morphologic changes in non-clonal SA: A Prominent cytoplasmic vacuolization in granulocytic precursors and in early erythroid precursors, with partial maturation arrest and significant dysgranulopoiesis in an infant with copper deficiency [A]. Dysgranulopoiesis with prominent hypogranulation and nuclear holes/defects in an adult with copper deficiency [B]; (Wright-Giemsa. ×500, 1000). |
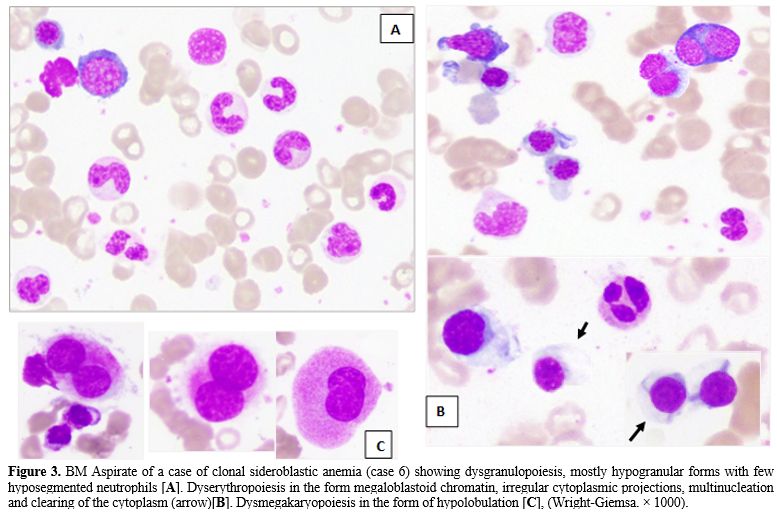 |
Figure 3. BM Aspirate of a case of clonal sideroblastic anemia (case 6) showing dysgranulopoiesis, mostly hypogranular forms with few hyposegmented neutrophils [A]. Dyserythropoiesis in the form megaloblastoid chromatin, irregular cytoplasmic projections, multinucleation and clearing of the cytoplasm (arrow)[B]. Dysmegakaryopoiesis in the form of hypolobulation [C], (Wright-Giemsa. × 1000). |
Clinically, not much difference between both groups regarding the need for blood transfusion. However, anemia was reversible in all cases with non-clonal SA, while none of the subjects with clonal SA had significant improvement (P=0.001). Vice versa, all of them with clonal SA had progressive disease and fatal course (100% vs. 0%, P=0.002). Due to the smaller number of cases, all comparative analyses were performed above as an exploratory statistical analysis; therefore, the derived statistical inferences might limit their conclusiveness and generalizability.
Acquired copper deficiency is a rare disorder associated with gastric bypass surgery or in cases with total parenteral nutrition or exclusively breastfed babies (as seen in case 1).[8]
In this patient, the prominent cytoplasmic vacuolization was the key to the diagnosis, confirmed by low serum copper. Furthermore, a positive therapeutic trial of copper (in high concentration) led to a significant improvement of the peripheral blood counts, which again dropped after copper supplements had been stopped. Finally, FCM analysis on BM aspirate of this baby showed an aberrant loss/down-regulation of CD33 on blasts, granulocytes, and monocytes, which is a rare occurrence of uncertain significance and might be a sign of dysmyelopoiesis.
In case number 2, the patient was initially misdiagnosed as MDS-RS based on the prominent multilineage dysplasia. However, no evidence of clonality was detected, with normal KT, negative SF3B-1, and no clinically significant variants identified using NGS. Additionally, it was observed that the CBC parameters improved significantly after hemodialysis. Copper serum level was then measured and found to be extremely low; hence, SA secondary to copper deficiency was concluded. Pancytopenia due to copper deficiency in a haemodialysis patient has been previously reported by Melero et al.[9] Furthermore, excessive zinc intake and copper deficiency induced SA/pancytopenia during maintenance haemodialysis has also been reported by Marumo A and his group.[10]
SA diagnosed during pregnancy is even a rarer event. In Case 3, the anemia was aggravated during pregnancies and normalized in between. This behaviour had pointed to possible vitamin/trace element deficiency owed to increase demand during pregnancy, especially since she had an associated B12 defect. Although CSA could not be entirely ruled out in this patient (as familial mutations were not done), however, the type of anemia (normocytic rather than microcytic), a lacking family history, absence of dysplasia, and detection of wild-type SF3B-1 gene, had all favoured an acquired cause.
Because of the crucial role of pyridoxal phosphate (the active form of vitamin B6) as an essential cofactor for ALAS2, a severe deficiency in vitamin B6 due to alcohol intoxication, increased demands, malnutrition/ malabsorption could lead to SA.[11]
It is worth noting that 4 out of 14 cases (28%) had an associated autoimmune disorder with DAT-positive hemolytic anemia found in two patients.
In cases (4 & 5), the exact cause of SA remained unrevealed, and there was no evidence of clonality detected. Interestingly, both patients had associated autoimmune disorder, with one patient having an adult Still's disease on hydroxychloroquine (case 4); in the other patient, SA was associated with DAT-positive autoimmune hemolytic anemia. We did not find a reported association between hydroxychloroquine and SA.
This association between SA and autoimmune disorders was rarely documented in the literature; however, a transient appearance of RS in the acute phase of secondary hemolytic anemia was reported by Wang and his group.[12]
Interestingly, an associated megaloblastic anemia was found in 6/15 cases (40%) of ASA; including 3 non-clonal cases and 3 cases associated with myeloid neoplasms.
AML with RS is rarely encountered and can be seen in de novo AML and secondary AML on top of MDS. We had a single case of a male patient diagnosed at a relatively younger age (37 years old) and considered the youngest among all groups of myeloid neoplasms with RS in our cohort. Martin-Cabrera et al.[13] recently reported that AML with RS shows a unique molecular signature straddling secondary AML and de novo AML.
In our cohort, NGS (including SF3B-1 mutation) was performed in 8 out of 15 cases, including 3/5 of the non-clonal cases. Within the group of clonal SA, clonality was established in 7/10 cases either by abnormal KT and/or detection of clinically significant variant using the NGS technique. Although molecular testing could not be performed in the remaining three cases (Case # 7, 12 and 15) due to lack of material, those cases had convincing evidence of MDS based on the striking morphologic findings and the aggressive disease course. TP53 mutation was detected in two cases in our cohort (cases 9 and 14). According to a recent large-scale genetic profiling study on MDS with RS, focusing on SF3B1-wild type (WT) patients, the group found that in SF3B1-wt patients, the most frequent mutation was TP53 at 61% (n = 92).[14]
The treatment of SA relies on the underlying aetiology but remains principally supportive with packed red cell transfusion for symptomatic patients, vitamin B6 supplementation, and iron chelation therapy for iron overload. Unlike SA due to myeloid neoplasms, the prognosis was favourable in all cases of non-neoplastic SA (in our series), with recovery of anemia either after replenishment of the deficient element (copper/pyridoxine) or spontaneous recovery in cases with idiopathic SA.
In conclusion, Although SA is uncommon and some forms are rare, it should be considered in patients with unexplained persistent anemia of any severity. It is sometimes difficult to distinguish congenital from acquired causes or to reach the exact cause of acquired SA and differentiate clonal from benign. The latter differentiation is crucial since acquired SA may have significant myelodysplasia, which could not be distinguished from MDS based solely on morphologic findings. Compared to the non-clonal group, the neoplastic SA presents in the elderly and tends to have lower WBC, higher blasts and RS, more frequent dysmegakaryopoiesis, and a more aggressive clinical course with fatal outcome.
Reaching a specific diagnosis requires a multiparametric approach relying on multiple factors, including the age of clinical onset, type of anemia (microcytic versus normocytic/macrocytic), an associated syndromic/dysmorphic feature, the degree of myelodysplasia, blasts percentage and most important the detection of clonal cytogenetics/ molecular genetic marker.