ACP should be considered in patients with unexplained atypical microcytic anemia untransfused without inflammation, low TS, and high ferritin levels. ACP diagnosis can be easily confirmed by low serum ceruloplasmin levels and is usually based on very low or undetectable serum CP levels and clinical, biochemical, or radiological signs of iron accumulation in the target organs.[1,4] In addition to these findings, biallelic pathogenic variants in CP are determined using molecular genetic tests in a proband (index case) to make a definitive diagnosis.[4,5] Early diagnosis and treatment are essential for preventing neurological complications of the disease, as once they occur, such complications are irreversible.[1,2,4]
We present the case of a 15-year-old girl with unexplained microcytic anemia, high ferritin levels, and low TS without inflammation.
The patient had anemia and growth retardation for approximately 5-6 years. Her parents were first-degree cousins. Her height was 148 cm (3%↓), and her body weight was 44 kg (3%↓). Her physical examination revealed pallor; however, other systemic functions were normal.
According to the laboratory results, the patient had mild hypochromic microcytic anemia, reduced TS, and hyperferritinemia (Table 1). "Atypical (Ferritin↑) microcytic anemia" is characterized by this biochemical triad, as opposed to the typical cases caused by iron deficiency wherein serum ferritin levels are consistently low.[1] A differential diagnosis could also be with iron-refractory anemia, usually acquired and often associated with gastrointestinal pathologies. However, a rare genetic form called iron deficiency anemia, iron-resistant iron deficiency anemia (IRIDA), also exists. In some pathological circumstances - congenital or acquired - hepcidin level increases, limiting the absorption of iron in the gastrointestinal tract and remobilizing and recycling iron, thereby reducing iron levels in plasma. Hence, conditions with high hepcidin levels are often underestimated as iron-refractory anemia, leading to inappropriate and unsuccessful treatments.[1,3] When evaluated in terms of differential diagnosis, the causes of hypochromic microcytic anemia include IRIDA, thalassemia (α and β), sideroblastic anemia, anemia of chronic disease (infection, cancer, inflammation, and renal disease), lead poisoning, hemoglobin E carriage, and severe malnutrition, were not detected in our patient. Hence, congenital iron metabolism disorders or copper deficiencies were considered the differential diagnoses of hypochromic microcytic anemia.[6,7]
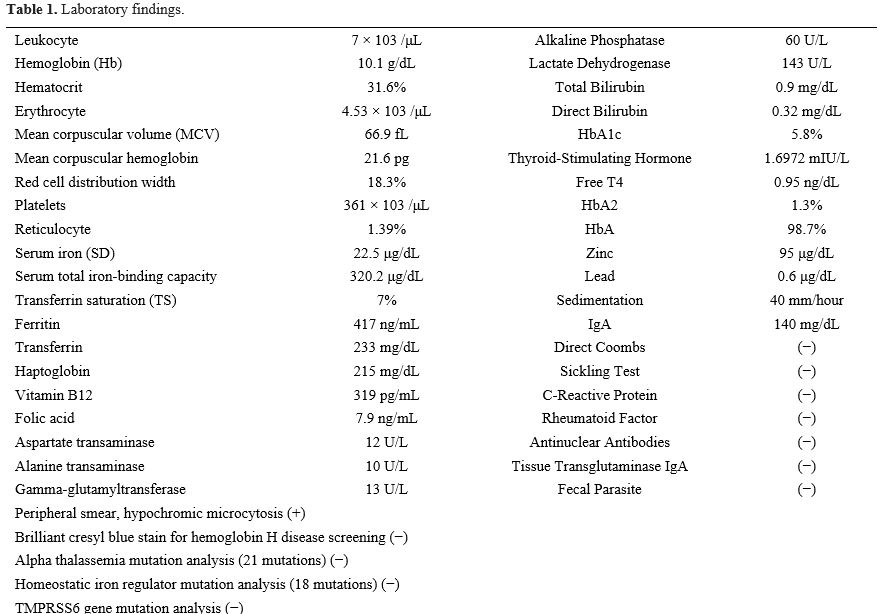 |
Table 1. Laboratory findings. |
Laboratory features of congenital iron metabolism disorders with iron deficiency and accumulation, which are considered causes of hypochromic microcytic anemia in differential diagnosis, are summarized in Table 2.
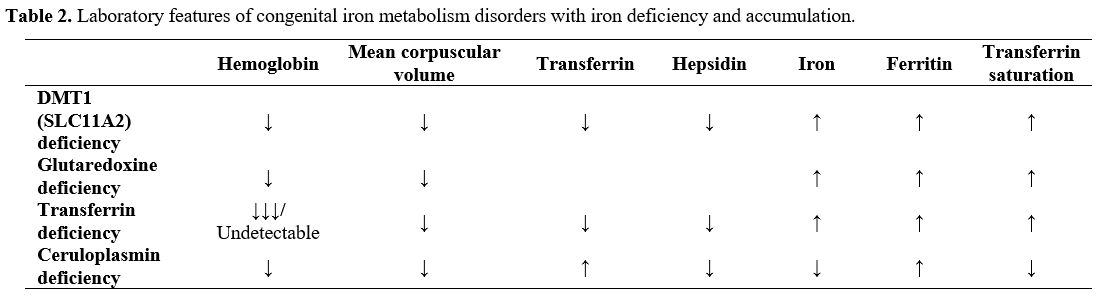 |
Table 2. Laboratory features of congenital iron metabolism disorders with iron deficiency and accumulation. |
The clinical characteristics of our patient were consistent with copper/ceruloplasmin deficiency inducing hyperferritinemia, low TS, low serum copper and ceruloplasmin levels, and normal urinary copper levels. Copper/ceruloplasmin deficiency can also be observed in ACP, heterozygous carriers of asymptomatic ceruloplasmin mutation, Wilson's disease, Menkes disease, or hypoproteinemias.[1]
The patient's laboratory results were consistent with ACP's diagnostic criteria. Molecular genetic analysis was performed to determine ACP diagnosis, and a homozygous mutation was found. Disease-related mutations in CP cause destruction or severe reduction of ceruloplasmin's ferroxidase activity. To date, 28 missense, 17 frameshift, 13 insertion, and 8 nonsense mutations have been identified.[5] A frameshift mutation was detected in our patient. Most pathogenic variants in CP are predicted to be loss-of-function mutations, which deteriorate or alter the protein stability of the copper binding sites. As most aceruloplasminemia cases are caused by homozygous mutations, investigating the consanguinity between parents is crucial. A milder disease course has been reported in some individuals with a single CP mutation (simple heterozygosity).[1,4]
Third-degree consanguinity (first cousin) was documented between our patient's parents, and the patient's parents and sister were heterozygous carriers of the mutation. The family members did not exhibit anemia; however, their serum copper and ceruloplasmin levels were lower than normal (Table 3).
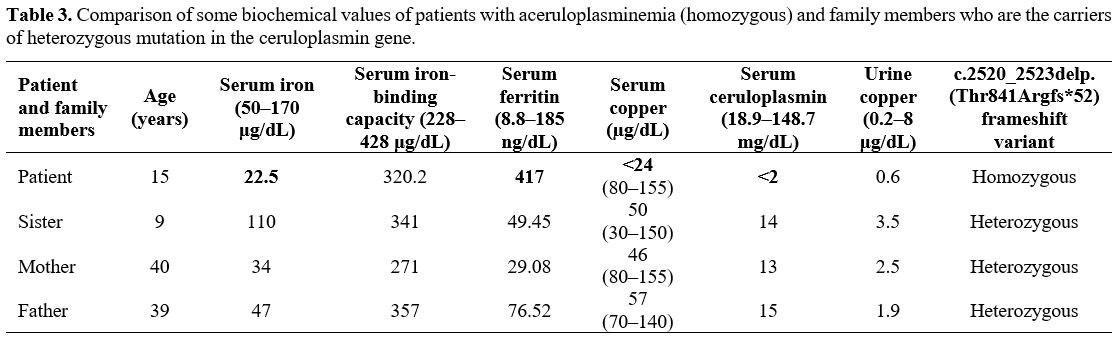 |
Table 3. Comparison of some biochemical values of patients with aceruloplasminemia (homozygous) and family members who are the carriers of heterozygous mutation in the ceruloplasmin gene. |
There was no difference in the hemogram values of other family members. Endocrine and neurology consultations performed for iron accumulation were normal. Fundus examination revealed subretinal deposits around the optic disk and macula. No significant degenerative findings were observed in the retina or choroid. Visual acuity was bilaterally complete, and intraocular pressure was bilaterally normotensive. Exams made to evidentiate iron overload were negative. Cardiac iron T2* value was a normal 21-22 Hz (47-48 ms) in measuring iron accumulation by magnetic resonance imaging (MRI). Moderate iron accumulation was detected in the liver (6.9–7.2 [0.2–1.6] mg/g iron). Brain MRI findings were normal.
However, the clinical presentation of ACP frequently includes cerebellar manifestations (dysarthria and ataxia) and involuntary movements (dystonia, chorea, and tremor), with onset typically occurring at 40–60 years. In contrast to this typical picture in Japanese patients with ACP, the age of onset tends to be earlier in Caucasians, with cognitive–psychiatric changes and extrapyramidal findings.[8,9] However, cognitive (apathy and memory loss) and behavioral changes have low specificity; hence, they are often underestimated.[4] Our patient exhibited no cerebellar symptoms or involuntary movements. There is no globally accepted treatment regimen for ACP. Iron chelating agents, which are highly effective in reducing liver iron deposition and can prevent further brain iron accumulation, are the first-choice drug; however, they are ineffective for patients with neurological damage that has already occurred.[4] According to expert opinion, the brain and visceral zinc concentrations in these patients decrease, zinc distribution exhibits a pattern opposite to that of iron, and zinc shows antioxidant activity; therefore, treatment with zinc-accompanied iron chelator in patients with ACP may be favorable in reducing iron deposition in the brain and other organs and in preventing or improving systemic and neurological symptoms.[5]
After the diagnosis of ACP, deferiprone 75 mg/kg/day three times a day was started for iron chelation. As a result, hemoglobin and serum iron levels increased, whereas serum iron-binding capacity and ferritin levels decreased. However, after three months of treatment, a decrease in hemoglobin levels was observed. Therefore, deferiprone was stopped. Zinc (50 mg/day orally) was initiated; subsequently, the patient's hemoglobin levels began to increase, and the patient's clinical condition improved.
Also, in patients with signs of iron overload, mild microcytic anemia was often observed in childhood as the earliest biochemical manifestation of ACP in both Japanese and non-Japanese cases. Still, ACP was rarely diagnosed in the early symptomatic period.[8,9] However, in our case, mild microcytic anemia, which our patient experienced for approximately 5-6 years, eventually led to a diagnosis of ACP as an early biochemical sign of the disease.
Other possible differential diagnoses are an iron transporter DMT1 (SLC11A2) deficiency, characterized by congenital hypochromic microcytic anemia; slow progressive hepatic iron accumulation; low hemoglobin, mean corpuscular volume (MCV), transferrin, and hepcidin levels; and high serum iron, ferritin, and TS levels; glutaredoxin (GLRX5) deficiency, WHICH present with pyridoxine-refractory sideroblastic anemia; hepatosplenomegaly; jaundice; cirrhosis; low hemoglobin and MCV levels; and high serum iron, ferritin, and TS levels. Moreover, ring sideroblasts are observed in the bone marrow of such patients. Congenital hypochromic microcytic anemia and hemochromatosis are present in a(hypo)transferrinemia cases wherein the transferrin levels are negligible/undetectable; hemoglobin, MCV, serum iron, and hepcidin levels are low; and ferritin and TS levels are high. Iron accumulation in the liver and brain and mild iron deficiency anemia is associated with ACP. In patients with ACP, ceruloplasmin is reduced/undetectable; MCV is low/normal; hemoglobin, serum iron, serum copper, TS, and hepcidin levels are low; transferrin and ferritin levels are high.[7,10]
In conclusion, unexplained atypical microcytic anemia without inflammation, low TS, and high ferritin suggest ACP, and low serum ceruloplasmin levels can easily confirm the diagnosis. The early diagnosis is crucial to prevent delayed treatment and irreversible neurological damage onset.