Elvira Pelosi, Germana Castelli and Ugo Testa.
Department of Oncology, Istituto Superiore di Sanità, Rome, Italy.
Correspondence to:
Ugo Testa. Department of Oncology, Istituto Superiore di Sanità, Rome, Italy. E-mail
ugo.testa@iss.it
Published: November 1, 2022
Received: September 17, 2022
Accepted: October 17, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022080 DOI
10.4084/MJHID.2022.080
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Despite
recent progress, acute myeloid leukemia (AML) remains a disease
associated with poor prognosis, particularly in older AML patients
unfit to tolerate intensive chemotherapy treatment. The development and
introduction in the therapy of Venetoclax (VEN), a potent BH3 mimetic
targeting the antiapoptotic protein BCL-2, inducing apoptosis of
leukemic cells, has shown to be a promising treatment for newly
diagnosed, relapsed, and refractory AML patients ineligible for
induction chemotherapy. Combination treatments using Ventoclax and a
hypomethylating agent (azacytidine or decitabine) or low-intensity
chemotherapy have shown in newly diagnosed patients variable response
rates, with highly responsive patients with NPM1, IDH1-IDH2, TET2 and RUNX1 mutations and with scarcely responsive patients with FLT3, TP53 and ASXL1
mutations, complex karyotypes, and secondary AMLs. Patients with
refractory/relapsing disease are less responsive to Venetoclax-based
regimens.
However, in the majority of patients, the responses have
only a limited duration, and the development of resistance is
frequently observed. Therefore, understanding the resistance mechanisms
is crucial for developing new strategies and identifying rational drug
combination regimens. In this context, two strategies seem to be
promising: (i) triplet therapies based on the combined administration
of Venetoclax, a hypomethylating agent (or low-dose chemotherapy), and
an agent targeting a specific genetic alteration of leukemic cells
(i.e., FLT3 inhibitors in FLT3-mutated
AMLs) or an altered signaling pathway; (ii) combination therapies based
on the administration of two BH3 mimetics (i.e., BCL-2 +MCL-1 mimetics)
and a hypomethylating agent.
|
Introduction
Apoptosis is an important biological process in health and disease and is regulated by BCL-2 family proteins.
BCL-2
was identified as an oncogene resulting from a translocation between
chromosomes 14 and 18 that promotes malignant lymphomagenesis. In the
early ’90s, BCL-2 was identified as a pro-survival protein preventing
apoptotic cell death.
These proteins exert either a pro-apoptotic
or anti-apoptotic effect, and their activity balance is crucial for
controlling cell viability.[1] The main activity of
these proteins consists in controlling the activation of caspases, the
proteolytic enzymes executioner of the apoptotic process.[1]
BCL-2 is a member of the anti-apoptotic protein family expressing
BCL-2-like homology domains 1-4, which includes, in addition to BCL-2,
BCL-XL, BCL-W, BCL2-A1, and MCL-1.[1] The BCL-2 family protein also
comprises some proteins with pro-apoptotic activity, including the
pro-apoptotic activators (BID, BIM, and PUMA), the pro-apoptotic
effectors (BAK and BAX), and the sensitizer effector (NOXA).[1]
All these proteins form the intrinsic apoptotic pathway. Intrinsic
apoptosis is executed in response to cellular damage and most
anti-cancer agents (Figure 1).
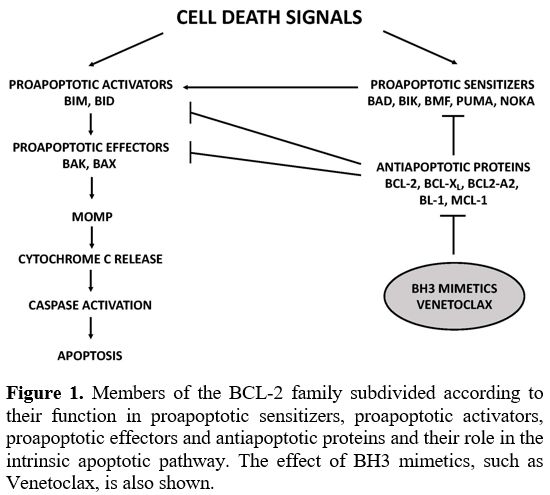 |
Figure
1. Members of the BCL-2 family subdivided according to their function
in proapoptotic sensitizers, proapoptotic activators, proapoptotic
effectors and antiapoptotic proteins and their role in the intrinsic
apoptotic pathway. The effect of BH3 mimetics, such as Venetoclax, is
also shown. |
BH
domains play a key role in controlling the activity of BCL-2 proteins
and the apoptotic process. BH3 domains are expressed by all the members
of the BCL-2 family; BAX and BAK proteins express all four BH domains;
the activator (BIM and BID) and sensitizer (NOXA) proteins contain only
BH3 domains. BH3 domain-mediated interactions between apoptotic and
anti-apoptotic BCL-2 family members play an essential role in the
control of apoptotic response: thus, the interaction of the sensitizer
and anti-apoptotic BCL-2 family members triggers apoptosis by enabling
activator proteins, not more bound to anti-apoptotic BCL-2 family
members, to interact with BAK/BAX effectors on the outer mitochondrial
membrane, resulting in the damage of this membrane with pore formation
and membrane permeabilization, the release of cytochrome C from
mitochondria, caspase activation and full induction of the apoptotic
machinery.[1]
Several BH3 mimetic drugs have been
synthesized, including venetoclax (VEN, ABT-199), navitoclax (ABT-263),
and ABT-737. These drugs selectively bind to the BH3 domain present on
anti-apoptotic proteins and, through this mechanism, induce the release
of bound pro-apoptotic proteins and apoptosis. Navitoclax binds to
BCL-2, BCL-XL, and BCL-W and, for this reason, induces in vivo
platelet lowering; this side effect is not observed with VEN that
selectively binds to BH3 expressed on BCL-2, thus sparing platelets.[2]
Preclinical
studies have supported the clinical evaluation of VEN as a potential
anti-leukemic drug. These studies have shown that AML bulk cells and
leukemic stem cells (LSCs) depend on BCL for their survival and BCL-2
inhibition causes cell death in AML cells.[3,4] In vitro
studies have shown that AML cell lines, primary patient AML samples,
and primary murine xenografts are very sensitive to treatment with VEN,
with induction of cell death.[3] Furthermore,
mitochondrial studies using BH3 profiling showed that VEN treatment
acts at the mitochondrion level, correlating with leukemic cell
cytotoxicity.[3] Other studies have provided evidence
that VEN's cytotoxic effect is also exerted at the level of LSCs, the
cells that initiate and maintain the leukemic process.[4]
In fact, it was shown that LSCs are present in a condition of
quiescence, with a low energy state and reactive oxygen species *ROS;
these cells are thus dependent on oxidative phosphorylation, whose
activity is dependent on oxidative phosphorylation and thus vulnerable
to BCL/2 inhibition using VEN.[4]
In initial
clinical studies, VEN was evaluated in monotherapy, but due to its
limited effects, it was evaluated in association with current
anti-leukemic treatments in subsequent studies.
Venetoclax in newly diagnosed AML
The
use of venetoclax in the treatment of newly diagnosed AMLs (ND-AMLs)
was mainly tested in patients with comorbidities precluding intensive
chemotherapy or in those older than 65-70 years. In these studies, VEN
was used in association with azacytidine (AZA) or decitabine (DEC), or
low-dose Ara C (LDAC). All these regimens have shown a good safety
profile and low 30-day mortality (Table 1).
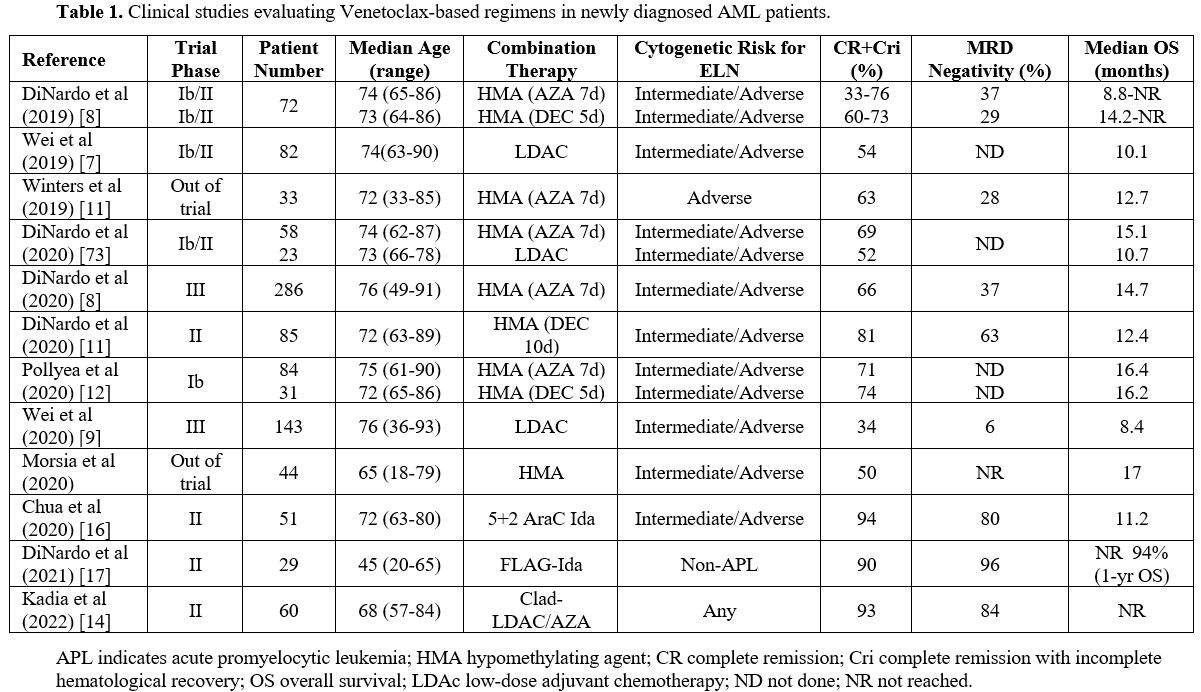 |
Table
1. Clinical studies evaluating Venetoclax-based regimens in newly diagnosed AML patients. |
Combination with hypomethylating agents.
DiNardo and coworkers have explored two groups of ND elderly AMLs, one
of 23 patients treated with 7-day AZA+VEN and the other of 22 patients
treated with 5-day DEC+VEN: in both treatment arms, a CR+Cri, with
incomplete count recovery (CRi), rate of 60% was observed.[5]
Subsequently, the same authors reported the exploration of a large
number of patients (145 ND-AML patients at least 65 years old). In this
group of patients, two VEN doses were explored, 400 mg and 800 mg,
showing better results for the 400 mg dosage at the level of safety
profile and therapeutic efficacy; using the 400 mg VEN dosage, a global
CR+CRi rate of 73% was observed; patients with high-risk cytogenetics
showed a CR+CRi rate of 60%. In addition, the median duration of CR+Cri
was 11.3 months, and the mean OS was 17.5 months for all patients and
was not reached for those treated with VEN at 400 mg.[6]
The
low-dose Ara-C (LDAC) regimen based on LDAC and VEN was explored in a
group of 82 older AML patients not eligible for IC (49% of these
patients had s-AML and 32% had poor-risk cytogenetic features): 54% of
these patients achieved a CR+CRi, with a median OS of 10.1 months and
DOR of 8.1 months.[7] In patients without previous
hypomethylating agents (HMA) exposure, CR+CRi was 62%, with a mean
overall survival of 13.5 months.[7]
Two
randomized clinical trials showed the superiority of the combined 7-day
AZA+VEN compared to 7-dayAZA+placebo and LDAC+VEN compared to
LDAC+placebo. Thus, a large phase III clinical trial (VIALE-A trial)
involved the study of 431 elderly AML patients randomized to receive
7-day AZA+VEN or 7-day AZA+placebo: CR+CRi rate was 66% vs. 28%, and
the median OS was 14.7 months vs. 9.6 months in 7-day AZA+VEN and 7-day
AZA+placebo, respectively, thus showing a consistent benefit deriving
from AZA+VEN administration compared to AZA alone.[8]
In the second trial, 211 elderly AML patients were randomized to
receive LDACc+VEN of LDAC+placebo: CR+CRi were 48% and 13% for LDAC+VEN
and LDAC+placebo, respectively; median OS was 7.2 months for LDAC+VEN
compared to 4.1 months for LDAC+placebo.[9]
Interestingly, 164 patients of the VIALE-A trial with CR+CRi were
explored for MRD status as assessed by multiparametric flow cytometry:
with a cut-off of <10-3, 41% of patients displayed an MRD-negative
condition, and 59% were MRD-positive; in MRD-negative patients after a
follow-up of 12 months DoR, EFS and OS were not reached, whereas in
MRD-positive patients DoR, EFS and OS were 81%, 83% and 94%.[10]
Winters
et al. have reported a “real-world” experience of VEN with AZA in 33
newly diagnosed AML patients; these patients received the same
treatment as another group of AML patients enrolled in phase I/II
study.[11] The CR+CRi rate was 63% for out-trial
patients, compared to 85% of the trial patients; the mOS was 381 days
for out-trial patients, compared to 880 days for trial patients.[11]
Prior exposure to hypomethylating agents was associated with poor
outcomes. On 14 patients out-trial, the MRD was evaluated after
treatment: 4/14 were MRD-negative and displayed sustained remission;
10/14 were MRD-positive, and 6 of these patients showed sustained
remission, while the 4 other patients relapsed.[11]
Pollyea
et al. reported the results of a phase Ib study of VEN with azacitidine
(84 patients) or decitabine (31 patients): the CR+Cri rate was 71% for
VEN+AZA and 74% for VEN+DEC; the median duration of CR/Cri was 21.9
months and 15.0 months, and the median OS was 16.4 months and 16.2
months, respectively.[12]
In order to improve
the rate and the duration of responses, more intensive treatments were
associated with VEN. DiNardo et al. have explored the safety and the
therapeutic impact of the administration of DEC 20mg/m2
for 10 days and VEN 400 mg daily for induction, followed by DEC for 5
days with oral VEN 400 mg for consolidation in a group of 70 elderly
ND-AML patients and 15 untreated s-AML patients: ORR was 89% and 80%
for ND-AML and s-AML patients, respectively; OS was 18.1 months for
ND-AML and 7.8 months for s-AML.[13] A more
comprehensive report on these patients, including 80 ND-AML and 20
untreated s-AML treated with 10-day DEC+VEN, explored the therapeutic
responses in genomic subgroups of patients: patients bearing NPM1, FLT3, IDH1/IDH2, TP53, RUNX1, N/KRAS mutations shower CR+CRi rates ranging from 70 to 88%; patients with ASXL1 and TP53 mutations displayed 55% and 50% of CR+CRi.[14] Median OS values for ND-AML patients were not-reached for NPM1-mutant, 29.6 months for IDH1/IDH2-mutant, 24.5 months for FLT3-mutant, 24.5 months for ASXL1-mutant, 16.1 for RUNX1-mutant, 12.1 months for N/KRAS-mutant and 5.4 months for TP53-mutant
AMLs. A propensity score-matched analysis of DEC10-VEN vs. intensive
chemotherapy stratified by risk of treatment-related mortality (TRM)
showed that DEC10-VEN offers better outcomes compared to intensive
chemotherapy in terms of CR+CRi rate, lower rate of relapse, and longer
overall survival.[15]
Combination with Reduced Intensity Regimens.
Kadia et al. have evaluated in a group of 60 older (≥60 years) AML
patients a peculiar therapeutic regimen based on VEN added to
cladribine (CLAD) plus LDAC, alternating with AZA; after treatment, 93%
of patients had CR+CRi, 84% were MRD-negative, and after 22 months of
follow-up the median OS and DFS were not reached.[16]
These results support the conclusion that VEN+CLAD/LDAC alternating
with VEN+AZA is an effective regimen in older or unfit patients with
ND-AML.
A recent study reported the initial evaluation of AZA+VEN
in combination with the anti-CD47 mAb magrolimab in a small cohort of
17 ND-AML patients older/unfit or high-risk (14/17).[17]
94% of treated patients achieved a CR+CRi condition, with 55% of MRD
negativity. Although these observations involve few patients, the
results observed in these patients are promising given their frequent TP53 mutant status (50% of cases) and high-risk condition.
Combination with Intensive Chemotherapy.
Other studies have explored the safety and efficacy of VEN administered
with intensive chemotherapy. In this context, the first study by Chua
et al. explored VEN in association with a modified intensive
chemotherapy protocol (CAVEAT, an attenuated 7+3 regimen consisting of
5 days of cytarabine and 2 days of idarubicin, 5+2) in 51 AML patients
with a median age of 72 years; the overall CR+CRi rate among both de
novo and secondary AML (sAML) patients was 72%; CR+CRi rate was 97% in
ND-AML.[18] After a median follow-up of about 2
years, mOS for the overall study population was 11.2 months; markedly
longer mOS was observed among de novo AMLs (31.3 months) compared to
sAMLs (6.1 months).[18] The safety profile for the
patients receiving up to 400 mg VEN was usually good, with a number of
infectious adverse events for their frequency and grade, expected for
AML patients of this age undergoing intensive chemotherapy treatment;
thus, these results showed that therapy with VEN in combination with
intensive chemotherapy is feasible in an elderly AML population.[18]
A phase I/II clinical study enrolled 29 ND-AML patients of a wide range
of ages, suitable for intensive chemotherapy, who were treated with
FLAG-IDA (fludarabine, cytarabine, G-CSF, and idarubicin) combined with
VEN with an ORR of 97%, 90% of CR+CRi, 96% of MRD negativity; 69% of
these patients proceeded to allo-HSCT.[19] A recent
study reported the results of an expanded cohort of ND-AML patients
enrolled in the FLAG-IDA+VEN study; the results on response rate, CR
rate, and MRD status confirmed those previously observed.[20] Estimated 24-months EFS and OS were 64% and 76%, respectively.[20]
A post-hoc propensity score-matched analysis of prospective clinical
trials in patients of the Texas University supported the conclusion
that VEN plus intensive chemotherapy improved event-free survival;
however, overall survival did not differ significantly compared to that
observed in patients treated with intensive chemotherapy alone.[21]
Other
studies have explored VEN in association with standard 7+3 induction
chemotherapy. In an initial study, Stone et al. reported the
preliminary results on 10 DN-AML patients treated with 7+3 chemotherapy
(cytarabine at days 1-7 and daunorubicin at days 2-4) In association
with VEN (400 mg was the maximum tolerated dose): the ORR was 100%, and
75% of the patients achieved MRD-negative remissions.[22,23]
Very recently, Wang et al. reported the results of phase II, a
single-arm trial enrolling 33 ND-AML patients aged 18-60 years treated
with 7+3 induction chemotherapy and VEN at 400 mg.[24]
After one cycle of therapy, a CR rate of 91% was observed; 97% of these
patients in CR had an MRD-negative status; after 11 months of
follow-up, 97% of OS and 1-year EFS was 72%.[24]
Recent
studies have explored the combination of VEN with CPX-351; CPX-351 is a
dual-drug liposomal encapsulation of cytarabine (ara-C) and
daunorubicin at 5:1 molar ratio that is approved for the treatment of
newly diagnosed therapy-related AML or AML with myelodysplasia-related
changes. Drug synergism/additivity in preclinical studies provided a
rationale for combining CPX-351 + VEN clinically. A first study based
on only 5 newly diagnosed AML patients with adverse prognosis showed a
CR/CRi rate of 80%; 80% of these patients were transitioned to
hematopoietic stem cell transplantation.[25] The
second study explored 31 patients with de novo AML, with a median age
of 74 years, predominantly with poor-risk disease; CR+CRi was observed
in 57 of patients; MRD-negativity was observed in 75% of patients who
achieved CR or CRi; survival data are not yet mature.[26]
Venetoclax in refractory/relapsing AMLs
About
sixty percent of newly diagnosed patients with AML receiving frontline
induction/consolidation chemotherapy achieve a complete response, but
30-40% of these patients relapse. Relapsed or refractory AMLs
(R/R-AMLs) remain a population with very adverse prognosis and
necessitate improved therapeutic options. The successful use of
Venetoclax as frontline treatment supported the exploration of its
possible use for the treatment also of R/R-AML patients (Table 2).
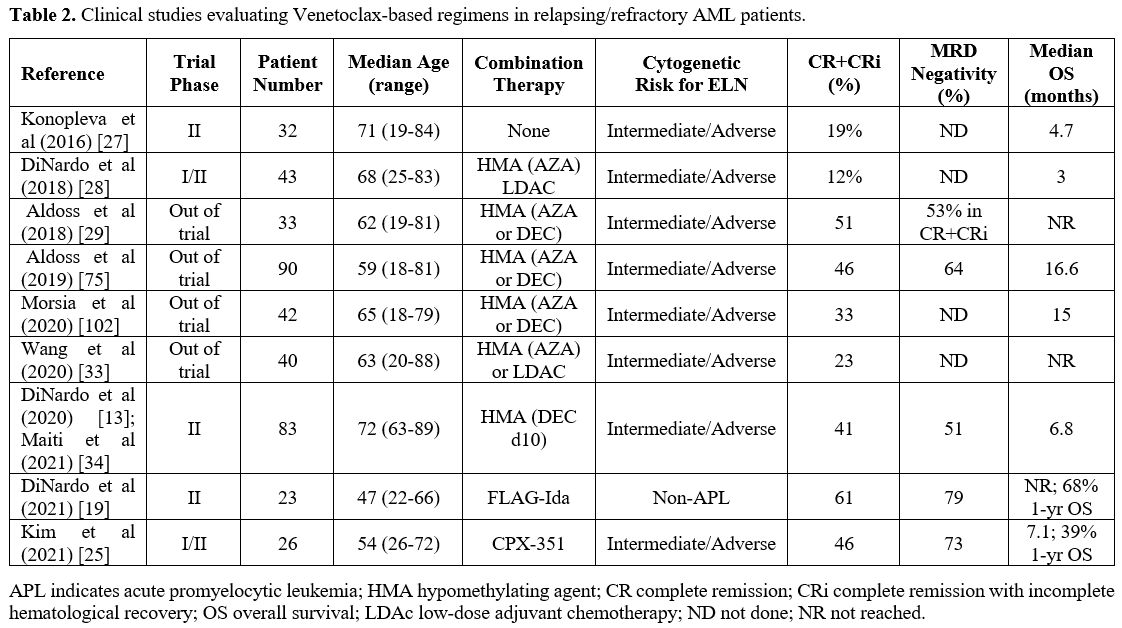 |
Table 2. Clinical studies evaluating Venetoclax-based regimens in relapsing/refractory AML patients. |
Combination with Hypomethylating Agents or Reduced-Intensity Chemotherapy.
In an initial phase II study, VEN was tested as a single agent in 32
R/R-AML patients and produced a limited CR+Cri rate of 19%.[27]
An
initial study by DiNardo and coworkers reported the clinical results of
treatment based on VEN+HMA or VEN+LDAC AML patients, using protocols
like those used in elderly de novo AML patients, in 39 R/R-AML patients, showing objective responses in 21% of cases, including patients with IDH1/2, RUNX1 and TP53 mutations.[28]
In another initial study, Aldoss et al. reported a “real-world”
analysis of 33 R/R-AML patients treated with either VEN+AZA or VEN+DEC,
reporting a CR+CRi rate of 51%.[29] 53% of CR+CRi responders were MRD-negative by multicolor flow cytometry.[29] Higher responses to the treatment were observed among patients with refractory de novo AMLs and therapy-related AMLs, compared to those with secondary AMLs.[29] The 1-year overall survival for all patients was 53% and was longer for patients with de novo than with secondary or therapy-related AMLs.[29]
Subsequent
studies have been performed using two different strategies: (i) some
studies explored standard VEN-based regimens using this agent in
combination with HMAs or with LDAC; (ii) other studies have evaluated
new VEN-based regimens. The first type of study involved, in most
instances, the limited experience of single centers and did not imply
controlled clinical trials. In this context, the study involving the
largest number of R/R-AML patients (86) was performed by Stahl et al.[30]
In this study, 86 R/R-AML patients were treated either with VEN+AZA or
with VEN+LDAC: VEN+AZA resulted in higher response rates than VEN+LDAC
(49% vs. 15%) and in a significantly longer OS (25 vs. 3.9 months). In
addition, mutations in NMP1 were associated with higher response rates,
whereas adverse cytogenetics and mutations in TP53, KRAS/NRAS and SF3B1 were associated with worse OS.[30]
Other
studies largely confirmed these findings. Labrador et al. reported the
results observed in 51 AML patients in the context of the PATHEMA group
who were treated with either VEN+AZA or VEN+DEC, or VEN+LDAC: the
frequency of responders (CR+PR) patients was higher for VEN+AZA (32%)
compared to VEN+DEC (13%) or VEN+LDAC (0%).[31] The patients enrolled in this study had very poor risk features and were heavily pre-treated.[31]
Feld et al. reported the results of their single-institution experience
in the treatment of 39 R/R-AML patients treated with VEN+HMA; 39% of
these patients achieved a CR/CRi, with an OS of 8.1 months; responders
to treatment were enriched for TET2, IDH1/IDH2 mutations, while non-responders were associated with FLT3 and RAS mutations.[32] Wang reported the results on 40 R/R AML patients treated with VEN-based therapy, showing 22% of CRs; patients with RUNX1
mutations showed a significantly longer OS; patients with
intermediate-risk cytogenetics had better outcomes compared to those
with adverse-risk cytogenetics.[33]
Combination with New Therapeutic Regimens.
The type 2 studies were based on the development of new therapeutic
regimens more appropriate for the treatment of a high-risk AML
population, such as R/R-AML. DiNardo et al. have developed a
therapeutic regimen involving a longer 10-day administration of
decitabine; it was hypothesized that VEN with 10-day DEC could lead to
an enhanced therapeutic response in both ND-AMLs and R/R-AMLs.[13] Thus, patients with R/R-AML received DEC 20mg/m2
for 10 days with oral VEN 400 mg daily for induction, followed by DEC
for 5 days with daily VEN consolidation. The overall response rate was
62%, with a median overall survival of 7.8 months and a duration of
response of 16.8 months.[13] The most significant rates of response and the longer OS were observed in patients with diploid cytogenetics, NPM1, and IDH1/IDH2 mutations.[14]
Maiti et al. recently reported the long-term outcomes of major genomic
subgroups of these RR-AML patients treated with DEC10-VEN: CR/CRi rates
in patients with mutations of NPM1 were 68%, of IDH1/IDH2 50%, of FLT3 42%, of RUNX1 45%, of TP53 30% and of KRAS/NRAS 26%; the longer OS was observed in patients with mutated IDH1/IDH2 (16.9 months), RUNX1 (13.7 months) and NPM1 (12.4 months), but shorter in patients with mutant ASXL1 (9.0 months), FLT3 (6.4 months), KRAS/NRAS (6.0 months) and TP53 (4.5 months).[15]
Maiti et al. have compared the outcomes of 64 R/R-AML patients treated
with DEC10-VEN to a cohort of 130 patients comparable for age and other
baseline characteristics treated with standard intensity chemotherapy
regimens commonly used for these patients: DEC10-VEN displayed
significantly higher responses compared to the IC cohort, including ORR
(60% vs. 36%) (MRD negativity assessed by multiparametric flow
cytometry (28% vs. 13%) and CR+CRi (19% vs. 6%). Multivariate analysis
supported a longer median event-free survival (5.7 vs. 4.5 months) and
median overall survival (6.8 months vs. 4.7 months) for DEC10+VEN
compared to IC.[34]
A phase I/II clinical study
enrolled 39 R/R-AML patients suitable for intensive chemotherapy
treatment who were treated with FLAG-IDA (fludarabine, cytarabine,
G-CSF, and idarubicin) combined with VEN: an overall response rate of
70-75% and a CR+CRi rate of 61-75% was observed.[19]
After a median follow-up of 12 months, median OS was not reached; 46%
of patients proceeded to allogeneic HSCT with a one-year survival
post-HSCT of 78%.[19] Wolach et al. have performed a
real-world analysis of 24 R/R-AML patients undergoing treatment with
FLAG-IDA+VEN and reported a CR+Cri rate of 72% (91% for patients
post-HSCT) and with an OS of 50% at 12 months.[35] A
registry-based study with FLAG-IDA+VEN corroborated the results
observed in the other studies with CR/CRi rate of 69%, MDR negativity
in 22% of patients, and 6-months OS of 10.5 months.[36]
A
recent study reported the preliminary results of a phase I/II clinical
trial based on the administration of AZA+VEN in association with an
anti-CD47 mAb (Magrolimab) in older ND-AMLs and in R/R-AMLs. Phase II
of the study involved 8 VEN-naïve R/R-AML patients and 13 VEN-treated
R/R-AML patients: in the former, a CR of 63% and an OS not reached were
observed; in the latter ones, a CR of 27% and an OS of 3.1 months were
observed.[17]
Ravandi and coworkers reported the
preliminary data on the safety and efficacy of combination therapy
based on DEC+VEN+ASTX727 (cytidine deaminase inhibitor cedazuridine) in
13 R/R-AML patients: ORR was 45%, with 30% of CR+CRi and an OS of 7.2
months.[37]
A recent study evaluated the safety
and the clinical efficacy of CPX-351 in combination with VEN, using an
approach similar to that adopted for newly diagnosed AML patients. 26
R/R-AMl patients were treated with CPX-351 and VEN, achieving a CR+CRi
rate of 46%, with an MRD negativity by flow cytometry of 75% and with
an mOS of 7.1 months (in the responding patients, the mOS was 26.9
months) and a 1-year OS of 39%.[25] In addition,
achieving measurable residual disease (MRD) absence was associated with
better OS in these patients, with an mOS of 26.9 months in MRD-negative
compared to 2.6 months in MRD-positive patients.[25]
The
treatment of IDH-mutant AMLs changed in the last years due to the
introduction in the therapy of IDH1 and IDH2-specific pharmacologic
inhibitors. A recent phase III trial in elderly IDH1-mutant
AML patients, who were ineligible for intensive induction chemotherapy,
showed a consistent clinical benefit deriving from the administration
of ivosidenib (an inhibitor of mutant IDH1) and azacytidine, with a mean OS of 24 months.[38]
Lachowicz et al. have performed a phase I/II clinical study involving
the administration of ivosidenib with VEN, with or without AZA, to a
group of either ND-AMLs or s-AMLs or R/R-AMLs.[39] The available results are relative to 8 R/R-AML patients: IDH1 mutation
clearance following treatment was achieved in 50% of these patients; at
24 months, 50% of these patients survived and MRD negativity correlated
with improved survival.[39] A clinical study reported the results of the clinical activity of EC10-VEN on a small cohort of 11 IDH2-mutant
R/R-AML patients, with an ORR of 82%, a CR-CRi rate of 54%,
MRD-negativity as assessed by Flow cytometry of 54% and by PCR of 36%;
after a follow-up of 21 months, 1-year OS was 59% and the mean overall
survival 14.7 months.[40] A preliminary report on 7 IDH2-mutant
R/R-AML patients showed the therapeutic efficacy of the triplet based
on the administration of AZA+VEN+Enasidenib: 86% of these patients
displayed CR+CRi, including those with prior exposure to AZA or
Enasidenib; the median OS was not reached, and the 1-year OS was 67%.[41]
Venetoclax as Maintenance Therapy
The
possible role of venetoclax in the maintenance therapy remains
undefined, and it remains unclear what is the optimal therapeutic
strategy for AML patients responding to venetoclax-based treatments.
In
this context, a recent study explored the possible consequences of
ceasing venetoclax-based therapy in responding patients. Thus, Chua et
al. have explored the effect of ceasing therapy in 13 patients ceasing
venetoclax administration in a condition of remission for a minimum of
12 months, compared to 16 comparable patients continuing therapy. The
median OS in the stop group of patients was 71.3 months, compared to
50.2 months in the group continuing the treatment.[42] During the observation period (>5 years), 46% and 69% of patients relapsed in the STOP and CONT groups, respectively.[42]
Although based on a few patients, these observations support the option
to stop venetoclax maintenance treatment after achieving at least 12
months of CR.
The benefit of allo-HSCT in patients achieving
response to venetoclax-based treatment is uncertain. In this context,
Pollyea et al. have explored a group of 119 ND-AML patients who
received AZA/VEN as initial therapy: 21 of these patients underwent
HSCT, while 31 additional patients were potentially eligible for HSCT
but deferred transplantation.[43] Median OS was significantly better among patients undergoing HSCT compared to those HSCT eligible not undergoing HSCT.[43]
Future studies will be required to define at the level of individual
AML patients the criteria required for selecting patients for ceasing
treatment or for HSCT based on prognostic disease criteria and response
evaluations.
Few studies have explored the possible use of VEN as
maintenance therapy. Kent et al. showed that VEN administration to 23
AML patients post-ASCT is tolerable without unexpected side effects.[44]
A larger number of patients and a longer follow-up are required to
assess the efficacy of VEN as maintenance therapy post-HSCT.[44]
A phase II study based on AZA+VEN administration for AML patients in CR
after intensive or low-dose chemotherapy as maintenance therapy: the
1-year OS was 93.8% in the intensive cohort and 53.% in the low-dose
cohort; of the seven patients with an MRD-positive status, 2 cleared
their MRD on AZA+VEN maintenance therapy; MRD-positive patients had a
median of molecular relapse-free survival (MRFs) of only 4 months,
compared to not reached for MRD-negative patients.[45,46]
These observations suggest that AZA/VEN maintenance is effective and
tolerable in patients not immediately eligible for HSCT after intensive
or low-dose chemotherapy induction.[45,46]
Several
ongoing trials are evaluating HMA-VEN after induction chemotherapy (NTC
04102020), after allo-SCT (NCT 04161885), and as MRD-directed therapy
after allo-SCT (NCT 04809181).
Outcomes of Selected AML Subtypes Following Venetoclax-Based Therapy
NPM1-mutated AMLs. NPM1 mutations occur in about 30% of AML patients; although typically associated with favorable prognosis, the beneficial impact of NPM1
mutations decreases in the presence of some co-mutations and with
increasing age in patients treated with intensive chemotherapy or with
HMAs. The studies carried out HMAs in elderly AML patients with NPM1 mutations
showed that the HMA+VEN drug combination is highly effective compared
with HMA alone or with intensive chemotherapy. Thus, a retrospective
analysis carried out by Lachowiez et al. showed in AML patients of age
>65 years treated with HMA+VEN a CR rate of 88%, 1-yr OS of 80%, and
mOS not reached; in patients treated with standard induction
chemotherapy, a CR rate of 56%, a 1-yr OS of 30% and mOS of 10.8 months
and in those treated with HMA alone a CR rate of 28%, a 1-yr OS of 12
months and an mOS of 4.8 months.[47]
In a
clinical study involving the treatment of ND AML patients with
DEC10-VEN, after a median follow-up of 25.4 months, treatment-naïve
NPM1-mutates AMLs displayed an mOS not reached, the highest compared to
other molecular subgroups.[13,14] In newly diagnosed
AML patients treated with VEN+AZA, a CR rate of 96% was observed,
compared to 89% with intensive chemotherapy and 36% with AZA alone; at
4 years, the OS with AZA+VEN was longer than with AZA alone or with
chemotherapy. [8,15]
The efficacy of VEN in NPM1-mutated AMLs is further supported by the retrospective analysis of 12 NPM1-mutated AMLs: 5 with molecular persistence of NPM1 mutations and 7 with molecular relapse/progression.[48]
All patients with molecular persistence achieved durable molecular CR
following treatment with VEN+low-intensity chemotherapy; 6/7 patients
with molecular relapse/progression achieved CR, MRD-negative, after 1-2
cycles of VEN+low-intensity chemotherapy.[48] These observations suggest a promising efficacy of VEN-based therapy also in high-risk NPM1-mutant AML patients.[48]
The
reasons for the high sensitivity of NPM1-mut AMLs to VEN-based
therapies remain to be determined. Studies performed in de novo elderly
AML patients have shown anti-leukemic activity in about 60-70% of these
patients, with NPM1-mut AMLs being the most responsive.[7]
One of the mechanisms through which AZA potentiates the pro-apoptotic
effects of VEN is related to its capacity to downregulate the
expression of MCL-1 and to enhance the expression of pro-apoptotic
proteins NOXA and PUMA, thus increasing the dependence of leukemic
cells on BCL-2 for their survival.[49]
Another
mechanism could be related to the capacity of VEN to target and kill
intensive metabolically. Previous studies have shown that LSCs are
characterized by a condition of quiescence and low energetic
metabolism, mainly maintained by a low rate of oxidative
phosphorylation, a process dependent on BCL-2, which can be inhibited
by VEN.[4] LSCs isolated from de novo AML patients are uniquely reliant on amino acid metabolism for oxidative phosphorylation and survival;[50] in cooperation with HMAs, VEN decreases amino acid uptake and, through this mechanism, induces LSC cytotoxicity.[51]
In contrast, LSCs isolated from relapsed AML patients are not reliant
on amino acid metabolism due to their ability to compensate through two
different mechanisms: increased fatty acid metabolism occurring as a
consequence of RAS pathway mutations[52] or increased
nicotinamide levels resulting from increased nicotinamide uptake via
NAMP transporter and synthesis through amino acid salvage pathway: the
increased nicotinamide metabolism activates both amino acid metabolism
and fatty acid oxidation driving oxidative phosphorylation.[53]
Interestingly the preclinical studies evaluating in vitro drug sensitivity have shown consistent responsiveness of NPM1-mut AML primary leukemic blasts to VEN: VEN displayed an IC50 of 289-486 nM vs. 4558-6539 nM for NPM1-mut and NPM1-WT specimens, respectively.[54] However, the sensitivity of NPM1-mut
AMLs to VEN was heterogeneous, with the FAB (French American British)
M1 class of these leukemias being sensitive and FAB M5B class (with
monocytic features) being resistant; furthermore, the
co-occurrence-in NPM1-mut AMLs of TET2 and PTPN11 mutations was associated with significantly reduced in vitro sensitivity to VEN.[54] Interestingly, this in vitro
screening also showed that RAD21-mut AMLs are highly sensitive to VEN;
this high sensitivity is extended to other mutations of cohesion genes,
such as SMC1A, SMC3, and STAG2.[54]
The high sensitivity of NPM1-mut AMLs could be related to the high expression of HOX genes in these AMLs: in fact, the high expression of HOXA genes is a marker of VEN sensitivity in primary AML samples.[55] These findings were recapitulated by the knockdown of the FOXM1
transcription factor (FOXM1 interacts with NPM1-mut protein and is
vehiculated to the cytoplasm by the mutant NPM1 protein), which induces
sensitization to VEN and a pattern of HOXA gene overexpression comparable to that observed in NPM1-mut AMLs.[56]
The elevated sensitivity of NPM1-mut AMLs could be related to the impaired mitochondrial function observed in these AMLs.[57]
Despite the good initial responses, a significant proportion of NPM1-mut
patients treated with VEN+HMA develop resistance and eventually
relapse. New drug combinations involving VEN with another drug that
could inhibit NPM1-mut have been identified to bypass this problem. One
of these approaches involves the association of VEN with a drug
inhibiting nuclear export. The selective inhibitors of nuclear export,
such as Selinexor and eltanexor, make part of a new class of molecules
that target exportin-1 (XPO1), a protein essential for the nuclear
export of major tumor suppressor proteins and of NPM1-mut protein.
Preclinical studies using Selinexor have shown that XOP1 inhibition
induces nuclear relocation of mutant NPM1
and reduces HOX gene expression, cell differentiation, and growth
arrest. Thus, there is a strong rationale for using these drugs to
develop new therapeutic strategies for treating NPM1-mut AMLs.[58] Studies in leukemic cell lines showed that VEN response was enhanced by selective inhibitors of nuclear export compounds.[59] However, patients with NPM1-mut
AMLs displayed only limited responses to Selinexor, which was also
associated with a consistent number of adverse events. These
observations support new clinical studies using eltanexor, a
second-generation XPO1 inhibitor, inducing fewer adverse events in
association with other anti-leukemic drugs, such as VEN.[60]
The
other approach implies the association of VEN with a menin inhibitor.
Increased expression of HOX genes is a specific feature of two AML
subsets, including MLL-rearranged and NPM1-mut
AMLs. Preclinical studies have shown that menin inhibitors inhibit AMLs
overexpressing HOX genes: these inhibitors block the interaction
between menin and MLL, thereby altering the binding of MLL to a subset
of its target genes, including MEIS1, a cofactor of HOX transcription
factors.[61-62] Phase III trials have shown that
monotherapy with menin inhibitors is well tolerated and has achieved
objective responses in patients previously treated with
relapsed/refractory AML harboring MLL rearrangements or NPM1 mutations. Furthermore, in mouse models of NPM1-FLT3-ITD
AMLs, VEN+menin inhibitor exerted a more potent anti-tumor effect
compared to menin inhibitor alone, eliminating leukemic cells,
including LSCs; these effects involve a decreased expression of BCL-2
and BCL-XL.[63] These results were confirmed in
another recent study, thus supporting the development of clinical
trials involving VEN+menin inhibitors for the treatment of MLL1-rearranged or NPM1-mut AMLs.[64]
IDH1-IDH2 mutated AMLs. After NPM1mutated AMLs, IDH1-2-mutated AMLs exhibit the most favorable outcomes following therapy with VEN+HMA. IDH1-mutant
AML patients in frontline therapy display a CR rate ranging from 75% to
100%, with a median overall survival (mOS) not reaching and 1-yr OS of
72%, while IDH2-mutant patients show a CR rate ranging from 75 to 86% with an mOS of 29.6 months.[65,66]
In the salvage setting, the outcomes of IDH1-2-mutated AML patients treated with VEN-HMA were inferior, with a CR rate of 33% and 1-yr mOS of 66%; in IDH1-mutant AMLs and a CR rate of 54%, with an mOS of 14.7 months in IDH2-mutant AMLs.[65]
Similarly, in the DE10-VEN study, a very high response rate was observed among patients with IDH1-2 mutations.[14]
In treatment-naïve patients, an ORR of 92%, with an mOS of 29.6 months
and mRFS not reached, and an MRD-negative status in 90% were observed;
in previously treated patients, the ORR was 71%, with a mOS of 16.9
months and a rate of MRD negativity of 70%.[14]
A pooled analysis of the results reported in the phase Ib study[6] and in the randomized phase III study[8] was recently published and showed for IDH1-2-mutated
AML patients a CR rate of 79% for VEN-AZA compared to 11% for AZA
alone, and a median duration of remission of 29.5 months for the
VEN-AZA group compared to 9.5 months for AZA alone, and a mOS of 24.5
months for VEN-AZA compared to 6.2 months for AZA alone.[67] In IDH1-1
wild-type AML patients, CR rates were 63% with VEN-AZA and 31% with AZA
alone, the mean duration of remission was 27.5 months for VEN-AZA and
10.3 months for AZA alone, and mOS 12.3 months for VEN-AZA and 10.1
months for AZA alone;[67] in IDH1-mutated patients, CR rates were 66.7% vs. 9.1% and mOS 15.2 months vs. 2.2 months; in IDH2-mutant patients, CR rates were 86% vs. 11% and mOS not reached for VEN-AZA vs. 11%.[67] IDH1-2 wild-type AML with poor cytogenetics treated with VEN-AZA had inferior outcomes compared to equivalent patients with IDH1-2 mutations; IDH1-2-mutated patients had a better outcome regardless of cytogenetic risk.[67]
Analysis of the genetic determinants affecting the response of IDH1-2-mutated AMLs to VEN-HMA supported the conclusion that IDH1-2 and NPM1 co-mutations tend to have favorable outcomes, whereas IDH1-1 and RAS pathway or TP53 co-mutations have lower outcomes.[66]
These
observations strongly supported the development of clinical studies
based on the combined administration of an HMA compound with VEN and a
selective IDH inhibitor. In this context, it is important to underline
that recent studies have supported both the safety and efficacy of
combining a hypomethylating agent with an IDH inhibitor. Thus, a recent
phase III trial in elderly IDH1-mutant
AML patients, who are ineligible for intensive induction chemotherapy,
showed a consistent clinical benefit deriving from the combined
administration of ivosidenib (an inhibitor of mutant IDH1) and azacitidine, with a mOS of 24 months.[68] Recently, Botton et al. presented the findings of the molecular analyses on newly diagnosed IDH1-mut
AML patients enrolled in the above-mentioned AGILE phase III study
comparing AZA+ivosidenib to AZA+placebo: 58 patients received AZA+IVO
and 62 AZA+placebo.[69] DNMT3A, SRSF2, and RUNX1 were the most frequent co-mutated genes in these patients; mutations in DNMT3A, RUNX1, SRSF2, and RTK pathway mutations were associated with improved outcomes.[69]
Furthermore, a phase Ib and II study showed that combination therapy
based on enosidenib plus azacitidine was well tolerated and
significantly improved overall response rates compared with AZA alone,
thus supporting that this therapeutic regimen may improve outcomes for
elderly AML patients with IDH2 mutant AML.[70]
It is important to note that it was shown that about 90% of newly diagnosed AML patients with IDH1-2
mutations achieve a MRD-negative status, as assessed by multiparametric
flow cytometry; however, only 52% of these patients achieved a
molecular MRD-negative condition as assessed by molecular evaluation of
residual IDH1-2 mutations.[66]
There is hope that the triplet therapy VEN+IDH inhibitor+ HMA may
augment the fraction of patients achieving a molecular MRD negativity.
Early
results of a phase Ib/II study explored the triplet combination with
ivosidenib, VEN with or without AZA in IDH1-mutated AML in newly
diagnosed and R/R patients, reporting CR rates of 100% and 67%,
respectively; the 1-yr overall survival in newly diagnosed AML was 100%
and that in R/R patients 50%.[71] Lachowiez et al.
recently reported the preliminary results of a phase Ib/II clinical
study involving the administration of ivosidenib with VEN, with or
without AZA, to a group of either de novo AMLs or s-AMLs or RR-AMLs; the available results were relative only to 8 R/R AML patients.[66[ IDH1
mutation clearance following treatment was achieved in 50% of these
patients; at 24 months, 50% of these patients survived, and
MRD-negativity correlated with improved survival.[72] In patients exhibiting massive leukemic cell lysis following treatment, the median OS was 42 months.[72] All patients relapsing after IDH1 mutant clearance showed no IDH1-mutant relapse.[72]
Venugopal and coworkers explored the safety and efficacy of enasidenib (a specific IDH2-mutant
inhibitor) and azacitidine in 26 AML patients: 7 newly diagnosed and 19
relapsed/refractory; the CR rate was 100% for newly diagnosed patients
and 58% for R/R AMLs.[73] Interestingly, 7 R/R
patients received the triplet ENA+AZA+Ven and showed a trend toward a
better mOS than those treated with ENA+AZA.[73]
Preliminary
results of a phase Ib/II clinical trial (Enaven-AML trial) explored
enasidenib in combination with VEN in a group of AML patients
previously treated with at least two lines of treatment and mostly with
relapsing or refractory disease.[74] A CR rate of 55% was observed, and all responders remained in remission during the study.[74]
Since IDH mutations induced inhibition of the TET2 enzyme, it seemed interesting to explore the effect of VEN-based therapies in TET2-mutated AMLs. The presence of TET2
mutations seems to be associated with high responsiveness to VEN,
particularly in relapsed/refractory AML patients, with complete
remission rates of up to 86% compared to 39% in patients with wild-type
TET2.[75]
The molecular mechanism responsible for the high responsiveness of IDH1/IDH2-mutated
AMLs to VEN remains largely undetermined and implies the dependency of
these leukemias on BCL-2. It was suggested that this marked sensitivity
to VEN could be related to a decrease of cytochrome C oxidase activity
induced by enhanced levels of 2-hydroxyglutarate (2HG) present in IDH2-mutated AMLs, lowering the threshold for VEN-induced apoptosis.[76]
FLT3-mutated AMLs. Several studies have retrospectively analyzed the response of FLT3-mutated
AMLs to VEN-based treatments. In an initial phase Ib study, treatment
with VEN and AZA or DEC showed a 72% CR rate, with a median duration of
remission (mDoR) of 11 months among FLT3-mutated AMLs.[6] These findings were validated and extended in the phase III VIALE-A trial showing that patients with FLT3
mutations had a better response to the treatment with AZA+VEN compared
to AZA+placebo: CR rates of 72.4% vs. 36.4% and mOS of 13.6 months vs.
8.6 months were observed.[12,77]
A recent study further analyzed FLT3-mutated AML patients included in these studies for their response to VEN-AZA-based therapy compared to AZA alone.[78]
The CR rates were 67% for the VEN-AZA group, compared to 36% for the
AZA alone group; the mDoR was 18.4 months and mOS was 14.7 months for
the VEN-AZA group to 13.4 months and 10.1 months, respectively for AZA
alone group.[78] In patients treated with VEN+AZA, the responses were higher for FLT3-TKD than FLT3-ITD-mutated patients: CR rates 77% vs. 63%; mOS 19.2 months vs. 9.2 months, respectively.[78]
A high rate of responses was observed among patients treated with ten-day decitabine with VEN (DEC10-VEN).[14]
In newly diagnosed AMLs, the CR rate to DEC10-VEN was 86%, with an
MRD-negativity of 80% and a mOS of 24.5 months; in previously treated
AML patients, the CR rate was 42%, with an MRD negativity of 70% and a
mOS of 6.4 months.[18] Among FLT3-NPM1 co-mutated patients not previously treated, CR rates were 88%, MRD negativity 92%, and mOS not reached; in FLT3-ITD co-mutated patients previously treated, CR rates were 56%, with MRD negativity in 86% and mOS of 12.4 months.[18]
It
is important to note that DiNardo et al. explored the molecular
patterns of response and treatment failure in 58 AML patients treated
with VEN+HMA and in 23 AML patients treated with VEN+LDAC.[79]
Primary and adaptive resistance to venetoclax was associated with the
enrichment or acquisition of leukemic clones activating signaling
pathways, such as FLT3 or RAS, or biallelically affecting the TP53 gene.[79]
Particularly, serial molecular analyses showed more frequently an
increased FLT3 clonal burden in some patients at the time of disease
progression and, more rarely, the acquisition of new FLT3-ITD mutations; furthermore, single-cell sequencing studies showed in some instances, the clearance of some FLT3-ITD-bearing subclones and the outgrowth of a resistant FLT3-ITD subclone.[79]
Studies in experimental models showed that FLT3-ITD causes dual
resistance to both VEN and LDAC that can be bypassed by the concomitant
addition of VEN and an FLT3 inhibitor.[79]
Studies
in preclinical models of FLT3-ITD AML showed that FLT3 inhibition
(using FLT3 inhibitors) combined with VEN showed a pronounced
anti-tumor activity and strongly supported clinical trials using this
drug combination.[80,81] In addition, BCL2 inhibitors
and FLT3 inhibitors synergize to induce the elimination of FLT3-ITD
mutated leukemic cells through BIM activation.[82]
At
the clinical level, the association of geltiritinib, a potent FLT3
inhibitor, with VEN showed a robust anti-leukemic activity with a CR
rate of 86%, molecular MRD clearance in 69% of responders, and mOS of
10.5 months.[83] Daver and coworkers reported the study of 56 R/R AML patients with FLT3 mutations (64% had received prior FLT3 inhibitor therapy) treated with VEN-geltiritinib.[84]
75% of these patients achieved a CR following treatment, which had a
similar rate in patients with or without prior FLT3 inhibitor therapy
(80% vs. 67%, respectively).[84] The mOS was 10.0 months, and molecular MRD negativity was reached in 60% of patients achieving a CR.[84]
Recent
studies have explored the triplet HMA, VEN, and FLT3 inhibitors. Thus,
in a recently published clinical study, a small cohort of older/unfit
patients with newly diagnosed FLT3
mutated AMLs was treated with a triplet regimen (HMA, VEN, and FLT3
inhibitor): 11 of the 12 patients treated with this therapeutic regimen
achieved CR, with MRD-negativity in 91% of these responding patients.[85]
In a more recent study, the same authors reported a retrospective
analysis of 87 older/unfit newly diagnosed AML patients with FLT3-mutated
AMLs treated with this triplet regimen (VEN+decitabine+FLT3 inhibitor)
compared to 60 similar patients treated with low-intensity chemotherapy
and an FLT3 inhibitor.[86] This study showed that
triplet therapy was associated with better clinical responses than
doublet therapy: CR rate 67% vs. 32%, molecular MRD negativity 96% vs.
54%. After a median follow-up of 24 months, patients receiving the
triplet regimen displayed a longer mOS than those treated with the
doublet regimen: not reached vs. 9.5 months, respectively.[86]
Another
recent study explored the triplet drug combination based on quizartinib
(a second-generation FLT3 inhibitor), VEN and DEC in newly diagnosed
and R/R patients with FLT3-ITD mutated AML.[87] Preclinical studies supported the rationale of the association of VEN with quizartinib.[80] In this preliminary report, the results on 13 R/R AML patients and 4 newly diagnosed AML patients were shown.[87]
In the 13 R/R AML patients (85% of these patients received prior
treatment with at least one FLT3 inhibitor), 69% of CRs were observed,
with 4/9 of these patients achieving a molecular MRD negativity; in the
four newly diagnosed AML patients, 100% of CRs were observed, with 100%
of molecular MRD negativity.[87] With a follow-up of 7.2 months, the mOS was not reached in the frontline cohort and was 7.1 months in the R/R AML cohort.[87]
A
phase I/II study explored the triplet combination based on azacitidine,
venetoclax and gilteritinib for FLT3-mutated AML patients with de novo (11 patients) or refractory/relapsing (15 patients) disease.[86]
In ND AML patients, 82% of CRs were observed, with 18% of patients
proceeding to HSCT; in R/R AML patients, 27% of CRs were observed.[88]
New drug combinations involving VEN are under preclinical evaluation for the therapy of FLT3-mutated AMLs. Thus, VEN synergizes with the AXL/MER tyrosine kinase inhibitor ONO-7475 in inducing the killing of FLT3-ITD-mutated AML cells.[81]
ONO-7475 even alone exerts an inhibitory effect on FLT3-ITD leukemic
cells, related to its capacity to inhibit ERK phosphorylation and
expression of the anti-apoptotic protein MCL1.[89] Importantly, the drug combination VEN+ONO-7475 is able to overcome VEN resistance of FLT3-ITD-mutated AML cells.[89]
Janssen and coworkers screened in vitro
654 anti-leukemic compounds in combination with VEN in 31 primary
samples of high-risk AMLs and observed that gilteritinib exhibited the
highest synergy with VEN in WT FLT3 AMLs.[90]
Importantly, the VEN+gilteritinib was active in inducing apoptosis of
leukemic cell lines and primary AML cells resistant to VEN+AZA.[90]
Mechanistically, the VEN+gilteritinib combination decreased
phosphorylation of ERK and GSK3B via combined inhibition of FLT3 and
AXL, mediating suppression of the MCL1 antiapoptotic protein through
induction of its proteasomal degradation.[90] These
observations support the evaluation of VEN+gilteritinib as a potential
therapeutic regimen for high-risk AML patients with FLT3 WT.
Potential resistance mechanisms of FLT3-mutated
AMLs may be represented by the inactivation of BAX expression mediated
by constitutive FLT3 activation and by enhanced expression of MCL-1
induced by FLT3-ITD. Two preclinical studies have explored mechanisms
driving the synergy between VEN and an FLT3 inhibitor.[82,91]
Thus, it was shown that treatment with A FLT3 inhibitor (midostaurin or
gilteritinib) alone or in combination with VEN elicited a
downmodulation of MCL-1 expression, seeming induced by simultaneous
suppression of multiple signaling pathways, including STAT5, RAS-MAPK
and PI3K-AKT.[82,90] The effect t
of the two drugs was complementary: gilteritinib treatment reduced the
binding of BIM to MCL-1 and increased the binding of BIM to BCL-2,
while VEN increased the binding of BIM to MCL-1 but inhibited the
binding of BIM to BCL-2.[82,91]
Importantly, co-treatment with VEN and gilteritinib increased the
binding of BIM to BAX without increasing the binding of BIM to other
BCL-2 anti-apoptotic proteins.[82] Thus, the
combination therapy decreased the binding of BIM to both BCL-2 and
MCL-1, liberating BIM for interaction with BAX and induction of
apoptosis.
AMLs with Spliceosome Mutations.
A retrospective analysis at a single institution (The University of
Texas, MD Anderson Cancer Center) analyzed 39 AML patients with
spliceosome mutations and 80 WT AML patients for these mutations and
treated with VEN in combination with hypomethylating agents.[92]
No significant difference in overall survival was observed between
patients with spliceosome mutations and those without these mutations
(35 vs. 14 months, respectively); 1-year overall survival was 63% in
the spliceosome cohort and 53% in the WT cohort.[92] For the various subtypes of spliceosome mutations, the OS for patients with SRSF2, SF3B1, and U2AF1 was not reached at 35 months and 8 months, respectively.[92] IDH2 mutations were enriched in patients with SRSF2 mutations and were associated with favorable outcomes; RAS mutations were enriched in patients with U2AF1 mutations and were associated with poor outcomes.[92]
TP53-mutated AMLs. The presence of TP53
mutations was associated with resistance to VEN. Preclinical studies
have shown that the TP53 apoptotic network is a main mediator of
resistance to BCL2 inhibition in AML cells.[93] In addition, knockout gene experiments have shown that the inactivation of genes such as TP53, BAX, and PMAIP1 results in venetoclax resistance in AML cell lines.[93]
The outcomes of AML patients with TP53
mutations are poor, with median overall survival in newly diagnosed AML
patients of about 5-10 months and in salvage settings of about 5
months.[79,94] However, in the DEC10+Ven trial, TP53-mutant
AML patients displayed a lower rate of CRs compared to patients without
these mutations (35% vs. 57%, respectively) and a lower rate of MRD
negativity (19% vs. 52%) and a markedly lower mOS (5.2 months vs. 19.4
months).[95]
Pollyea et al. recently reported the retrospective analysis of the high-risk AML patients reported in the phase Ib[6] and phase III[8]
studies involving VEN+AZA administration to older, newly diagnosed AML
patients. Particularly, the outcomes of poor risk cytogenetics+TP53-mut AML patients were compared to those of poor risk cytogenetics+TP53-WT: in poor risk cytogenetics+TP53-mut patients, VEN+AZA improved remission rates but not DoR and mOS compared to AZA alone; in poor risk cytogenetics+TP53-WT
patients a higher remission rates and longer DoR and mOS than AZA
alone, with outcomes similar to those observed in intermediate-risk AML
patients undergoing similar treatment were found.[96] These observations support the conclusion that among high-risk AML patients, those TP53-WT exhibit a better benefit than those TP53-mut following treatment with VEN+HMA.
In
a retrospective analysis performed on 81 AML patients treated with
VEN+HMA or VEN+LDAC, none of the 18 patients with TP53 mutations
displayed durable remission; some displayed primary resistance, and
others rapidly relapsed after an initial remission.[79] Individual serial molecular analyses of some of these patients showed an expansion of the size of the TP53-mutant clones with biallelic TP53 defects under therapeutic pressure; polyclonal selection of clones with biallelic TP53 mutation was observed at relapse, with the appearance also of additional TP53 variants.[79]
Very
interestingly, an ongoing clinical trial involving the administration
of VEN+AZA+Magrolimab (anti-CD47 mAb) to older/unfit AML patients
reported in 7 newly diagnosed TP53-mutated AML patients a CR rate of 86%, with MRD negativity in 57% of cases and complete cytogenetic response in 3 patients.[21]
Preliminary results on a very limited number of TP53-mutated AML patients showed that weekly VEN with low-dose DEC results in a high rate of clinical and molecular responses.[97] However, these observations need to be confirmed on a larger cohort of TP53-mut AML patients.
RUNX1-mutated AMLs. RUNX1-mutated
AMLs represent a particular subtype of AMLs (about 10% of
newly-diagnosed AMLs), being almost exclusive of AMLs with recurrent
genetic alterations.[98] These leukemias frequently co-occur with genetic mutations involving epigenetic modifiers, such as ASXL1, IDH2, KMT2A, and EZH2, components of the spliceosome complex, such as SRSF2 and SF3B1, STAG2, PHF6 and BCOR;
these AMLs usually have an immature phenotype and frequently are sAMLs
evolving from MDS.[98] Since a significant proportion of RUNX1-mutated AMLs evolves from a pre-existing MDS syndrome, it is fundamental to distinguish de novo cases from those evolving from MDS (sAML). The analysis of de novo RUNX1-mutated AMLs showed that these AMLs, compared to RUNX1-WT AMLs, displayed a higher frequency of SRSF2 and ASXL1 mutations of normal karyotype and absent NPM1 mutations.[99] De novo RUNX1-mutated AMLs showed an overall survival similar to that observed for RUNX1-WT AMLs, thus indicating that the poor prognosis of RUNX1-mutated AMLs is not due to the mutation itself but is attributable to pre-existent MDS.[99]
Few studies explored the response of RUNX1-mutated
AMLs to VEN+HMA as frontline therapy. DiNardo et al. reported in a
retrospective analysis that 33% of RUNX1-mutated AML patients exhibit
durable remission after VEN-HMA therapy, 13% remission then relapse,
and 45% primary resistance, thus supporting the existence of a
consistent heterogeneity of these leukemias to frontline therapy with
VEN+HMA.[79] Cherry and coworkers have retrospectively analyzed 143 de novo AMLs who received VEN-AZA and 149 who received intensive chemotherapy treatment; the presence of RUNX1 mutations in these patients was associated with better outcomes for VEN-AZA compared to intensive chemotherapy.[100] The benefit deriving from the VEN-AZA regimen over intensive chemotherapy was particularly evident for patients with RUNX1 mutation and an age >65 years.[100] Venogopal et al. have retrospectively analyzed 907 AML patients, including 137 patients with newly diagnosed mutRUNX1
AML who underwent first-line treatment based either on intensive
chemotherapy (IC), low-intensity therapy (LIT) or LIT+VEN: there was no
significant difference in outcomes between RUNX1mut and RUNX1wt AMLs, regardless of therapy received; among patients who received LIT+VEN there was a trend towards better survival with mutRUNX1 AML compared to those without mutRUNX1 (25.1 vs. 11.3 months of overall survival with a 2-year overall survival of 54% vs. 33%).[101] Furthermore, in patients without other adverse-risk cyto-molecular features, the presence of mutRUNX1 conferred inferior overall survival in patients who received IC or LIT but not in those treated with LIT+VEN.[101]
In addition to the studies on de novo RUNX1-mutated
AMLs, studies on refractory relapsing patients support good
responsiveness to VEN+HMA. Wang et al. explored the factors predictive
for response among 40 relapsing/refractory AML patients treated with
VEN-based regimens: patients harboring NPM1, RUNX1, or SRSF2 mutations seemed to have higher complete remission rates, and mOS was significantly longer in RUNX1-mutated AMLs.[29]
DiNardo reported a retrospective analysis in 43 refractory/relapsing
AML patients treated with VEN-based regimens and observed 50% of
clinical responses among RUNX1-mutated AML patients; interestingly, the
TP53-mutated patients who responded to treatment had concurrent RUNX1
mutations and, similarly, of the 15% of responding patients with
adverse cytogenetics, all had concurrent RUNX1 mutations.[25] Other studies in relapsing/refractory AML patients bearing RUNX1 mutations have shown a rate of objective responses from 35% to 75%.[102]
The sensitivity of some AMLs bearing RUNX1 mutations to VEN may be related to the differentiation stages of these AMLs and to some peculiar effects induced by RUNX1 mutations at the level of the hematopoietic stem cell compartment. Mutations in RUNX1
reduced ribosome biogenesis, metabolism, and sensitivity for induction
of apoptosis in hematopoietic stem cells, thus creating resistance to
endogenous and genotoxic stress.[103] The impaired ribosomal biogenesis is a condition that renders RUNX1-mutated
AMLs more sensitive to the protein translational inhibitor
hemaharringtonine (omacetaxine) and to VEN: hemaharringtonine treatment
reduced the levels of c-Myc, c-Myb, MCL-1, and BCL-XL and,
consequently, synergized with VEN in inducing apoptosis of AML cells
expressing mutant RUNX1.[104] This combination treatment improved the survival of immunodepleted mice engrafted with AML cells bearing mutant RUNX1.[104] The sensitivity of RUNX1-mutated
AMLs to VEN could also be related to their arrest at an early stage of
hematopoietic differentiation. In fact, AMLs harboring RUNX1
mutations or inv(3) are among the different AMLs blocked at the
earliest stage of hematopoietic stem cell/progenitor-like
differentiation.[105]
Secondary AMLs.
Secondary AMLs (sAMLs) derive from the leukemic transformation of
preceding myeloid neoplasia, either a myelodysplastic syndrome (MDS) or
of myeloproliferative neoplasms (primary myelofibrosis, polycythemia
vera, or essential thrombocytosis). These AMLs are poorly responsive to
standard treatments and have poor prognosis.
Unfortunately, the
studies with VEN-based therapies have also shown a limited response in
patients with sAML. A single-center evaluated VEN-based combinations
(either with HMA or with chemotherapy) in 14 ND and 17 R/R patients
developing AML post-myeloproliferative neoplasms.[100]
In frontline patients, CRs were observed in 54% of patients, while no
objective responses were observed in R/R patients; the median duration
of response among newly diagnosed patients was 6.4 months.[106]
Data pooled from the VIALE-A study showed that patients with sAML
evolving from preceding MDS or MPN demonstrated superior response rates
and overall survival when treated with AZA+VEN compared to AZA alone:
CRs 66% vs. 27%; mDoR 15.9 vs. 10.1 months.[107]
Short
et al. have reported the retrospective analysis of 562 patients who
developed AML from preceding myelodysplastic syndrome (MDS) or chronic
myelomonocytic leukemia (CMML); these patients were stratified
according to frontline therapy: intensive chemotherapy (IC, 271
patients), low-intensity chemotherapy without VEN (LIT, 237 patients)
and VEN+HMA (54 patients).[102] Compared to IC or LIT, VEN+HMA induced a higher CR rate (39% vs. 25%) and a better overall survival (1-year OS 34% vs. 17%).[107]
Importantly, the benefit deriving from VEN+HMA treatment was restricted
to patients with non-adverse karyotype, with a mOS of 13.7 months and
1-tear OS of 54%.[108] In addition, patients who
underwent subsequent hematopoietic stem cell transplantation displayed
a superior 3-year OS compared to those not transplanted (33% vs. 8%).[108]
A
recent study explored the stem cell architecture of MDSs progressing to
AMLs and identified some properties of myelodysplastic cells predicting
response to VEN.[109] The bone marrow samples of one
group of MDS patients (52% of total) displayed an abnormal
differentiation pattern characterized by increased frequency of common
myeloid progenitors (CMP) and the other group by increased frequency of
granulo-monocytic progenitor (GMP); this two MDS differentiation
patterns did not derive from the expansion of either the CMP and GMP
populations but were the consequence of the marked decrease of the
frequency of the other two respective progenitor populations: GMPs and
megakaryocytic progenitors (MEPs) in CMP-pattern MDS and CMPs and MEPs
in GMP-pattern MDSs.[109] At the HSC level, the CMP
pattern was associated with an expansion of LT-HSCs and MPPs, while the
GMP pattern was associated with an expansion of lymphoid-primed
multipotent progenitors (LMPPs). These two MDS architectures are driven
by different genetic alterations: TP53 and EZH2 mutations are significantly associated with the CMP pattern, while RUNX1, DNMT3A, BCOR, and STAG2 mutations are enriched with the GMP pattern.[109]
These two MDS patterns are maintained in MDS patients undergoing
treatment with HMSs alone. Importantly, during disease progression to
AML, these two different MDS patterns undergo the expansion of distinct
stem cells that activate specific survival pathways: the BCL-2 pathway
in the CMP pattern and the nuclear factor-kappa B-mediated survival in
the GMP pattern; in line with these findings, VEN-based therapy
selectively targets HSCs from CMP-pattern MDS at blast progression
after HMA therapy failure.[109]
Interestingly,
a recent report showed a high response rate in 44 patients with MDS
undergoing treatment with VEN+HMA, with an objective response rate of
75% in HMA-naïve patients, 62% in previously HMA-exposed patients, and
44% in patients after HMA failure.[110] Importantly,
this treatment also led to high allogeneic stem cell transplantation
rates performed in 62% of responding patients.[110] Factors associated with poor overall survival were represented by those previously identified in AML patients, including TP53 mutations and complex karyotypes.[110]
Safety Profile of Venetoclax
The
numerous studies carried out using VEN have allowed to evaluate of the
safety profile of this drug in the various drug combinations in which
it was used, showing that VEN administration is clinically feasible
when administered in combination with hypomethylating agents (AZA or
DEC), low-intensity chemotherapy, intensive chemotherapy and in the
context of “triplet” regimens in which this drug is administered in
combination with low-intensity chemotherapy or a hypomethylating agent
and a drug molecular targeting a leukemic genetic abnormality (i.e., an
IDH inhibitor in an IDH-mutated AML.[111-112]
The most relevant information concerning the safety profile of VEN derives from the two phase III studies discussed above.[8,9]
The VIALE-A study comparing VEN+AZA to AZA+placebo showed that the
VEN+AZA safety profile was consistent with the side-effect profiles of
these two drugs when used in monotherapy, and the observed adverse
events are those expected in a population of elderly AML patients.[8]
The most common adverse events in the two groups of patients were
hematologic and gastrointestinal, with a higher frequency of
neutropenia and febrile neutropenia in the group of patients treated
with VEN+AZA; furthermore, a higher incidence of dose interruptions but
not the discontinuation of treatment or reduction in doses, to allow
hematological recovery in patients treated with VEN+AZA.[8]
These results supported the conclusion that VEN+AZA administration is
feasible in a population of older AML patients and that monitoring and
management of myelosuppression are important clinical issues in AML
patients treated with VEN+AZA.[8] The findings of the
other phase III study involving the comparison of VEN-LDAC to
LDAC+placebo confirmed the findings of the VIALE study, showing that
among the hematological toxicities, only neutropenia but not febrile
neutropenia, thrombocytopenia and anemia, were more frequent in
VEN+LDAC patients compared to LDAC+placebo patients.[9]
Tumor
lysis syndrome (TLs) is observed in some AMl patients treated with
VEN-based regimens due to rapid and massive lysis of tumor cells. The
incidence of laboratory TLS was reported ranging from 1% to 6% among
patients undergoing treatments with lower-intensity VEN-based regimens
and from 0% to 6% in those treated with VEN+chemotherapy; however, the
incidence of clinical TLS was lower, ranging from 0% to 2.7% and this
is seemingly due to the use in clinics of therapeutic measures to
reduce WBC count less than 10x109 before to start the therapy with VEN combinations.[113]
These estimates of TLS were based on the evaluation of patients treated
in the context of clinical trials; however, for patients treated
outside of clinical trials, the estimate of the frequency of TLS is
around 5%.[114]
The analysis of AML patients
treated with VEN+chemotherapy regimens, such as FLG-IDA or CLIA,
resulted in similar myelosuppression compared to other intensive
chemotherapy treatments used in AML patients and required standard
antimicrobial prophylaxis.[112]
The good
tolerability of VEN-based regimens is also supported by two recent case
reports in extremely vulnerable patients with concurrent COVID-19
infection and AML: one patient with concurrent severe COVID-19
pneumonia and AML was first treated for infectious pneumonia and when
the patient’s conditions related to pneumonia improved was treated with
VEN+AZA achieving complete remission and was now potentially available
for hematopoietic stem cell transplantation;[115]
the other patients with COVID-19 infection had concurrent t-AML evolved
from chronic myelomonocytic leukemia, achieving complete remission when
sequentially treated with VEN+AZA, with the abrogation of both mutant
clones associated with AML evolution and pre-existing to leukemia
progression.[116]
Venetoclax Metabolism and Drug Interactions
Venetoclax
is primarily metabolized by cytochrome P450 isoform 3A4 (CYP3A4) and is
predominantly cleared by the liver, as shown by in vitro studies. In fact, in vitro studies of drug-drug interaction with ketoconazole, Posaconazole, and rifampin have supported the metabolism of VEN by CYP3A4.[117-118] Studies of drug metabolism in normal volunteers using a single dose of [14]-Venetoclax
showed that all the administered drug was excreted with feces, with
only a minimal contribution (0.1%) of the urinary tract; the extent of
drug absorption was around 65%.[119] VEN was
primarily cleaved by hepatic metabolism (66% of the administered dose);
33% of the administered drug was recovered as the parent drug and its
nitro reduction metabolite M30; M27 is a major drug metabolite and is
primarily formed by CYP3A4.[119]
The
pharmacokinetics of Ven was characterized in chronic lymphocytic
leukemia and lymphoma patients after a single oral dose, with VEN
plasma concentrations peaking at 6 to 8 hours after administration and
with a terminal phase elimination half-life of approximately 19 hours.[120-121]
Venetoclax
is a substrate of CYP3A4; antifungal agents used to prevent systemic
fungal infections in neutropenic hematologic patients are inhibitors of
CYP3A4: Posaconazole or voriconazole are strong inhibitors, while
isavuconazole or fluconazole are moderate CYP3A4 inhibitors.
Pharmacokinetic studies have shown that the concomitant administration
of an antifungal agent, such as posaconazole, with VEN, requires a VEN
dose reduction by at least 75%; both the 50- and 100-mg VEN doses
administered with Posaconazole were well tolerated, in spite the 400-mg
that represents the optimal VEN dose without concomitant antifungal
agents.[122] Using robust pharmacokinetic models for
the drug Posaconazole, it was estimated that the recommended dose of
VEN is 70 mg in the presence of Posaconazole up to 500 mg doses.[123]
The
clinical experience showed that the combination of VEN and antifungal
azoles results in prolonged cytopenias, namely, thrombocytopenia,
compared to using VEN without an azole.[124-125]
However, this effect did not result in higher rates of febrile
neutropenia, infections, or duration of hospitalization, thus
indicating that the concomitant use of VEN and antifungal azoles
represents a clinically safe and effective therapeutic regimen, after
adjustment of VEN dosage.[124-125]
Interestingly,
grapefruit, star fruit, and oranges can potentially increase VEN plasma
concentrations if taken concomitantly; this effect is related to the
capacity of these fruits to act as moderate CYP3A4 inhibitors.
According to these findings, it is recommended to avoid eating these
fruits during VEN-based treatment.
Recent studies suggest that
antifungal agents enhance VEN-AUC not only through inhibition of CYP3A4
but also through inhibition of the transporter OATP1B1, involved in the
elimination of VEN.[126]
Venetoclax Resistance
The most consistent limitation of the therapy of AMLs with VEN is the short duration and the development of resistance.[127]
Therefore, understanding the mechanisms of resistance to this drug is
of fundamental importance for the development of new strategies able to
bypass these therapeutic blocks and for the definition of new drug
combinations.[127] Various mechanisms of resistance
to VEN-based regimens have been identified in AML cells. However, the
two most important are represented by dependencies on alternative
anti-apoptotic BCL-2 family members and the selection of activating
kinase mutations.[127]
The mechanism related to
the development of dependencies on alternative anti-apoptotic BCL-2
proteins seems to be particularly relevant. These alternative
dependencies include different anti-apoptotic mediators, such as
BCL-2-A1, MCL-1, and BCL-XL and are involved in primary and adaptive
VEN resistance mechanisms (Figure 2).
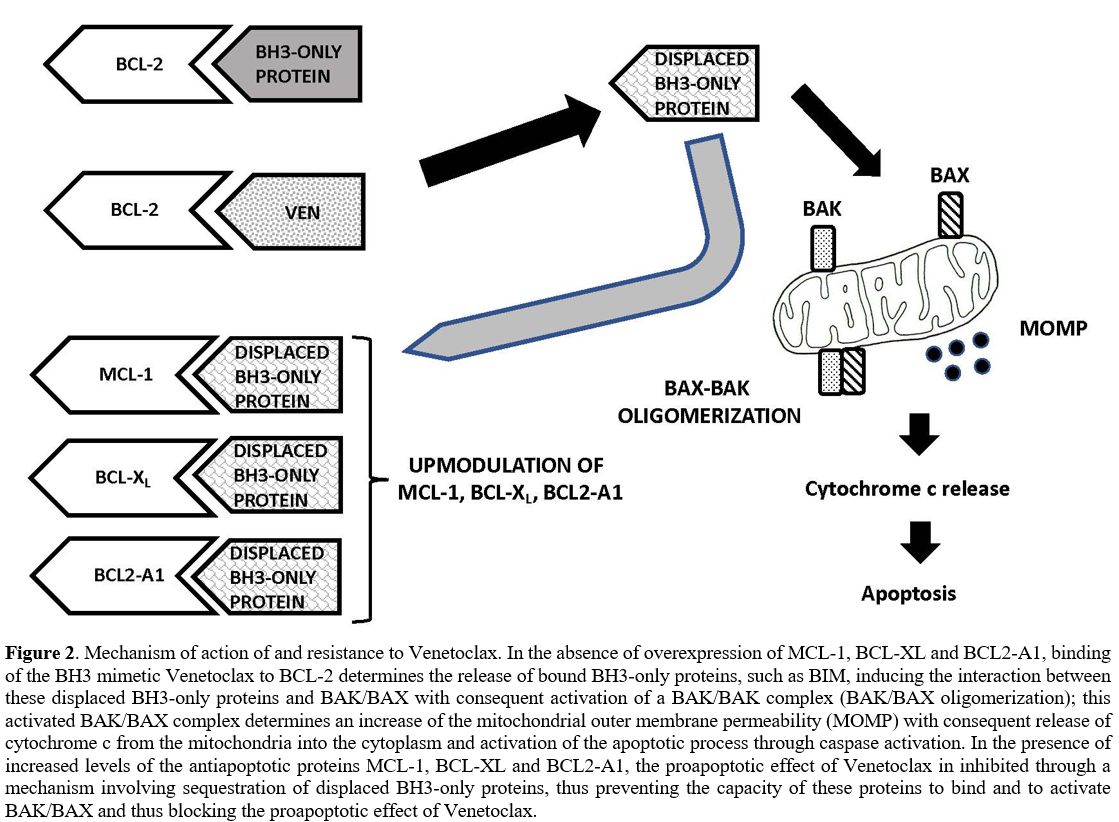 |
Figure 2. Mechanism of
action of and resistance to Venetoclax. In the absence of
overexpression of MCL-1, BCL-XL and BCL2-A1, binding of the BH3 mimetic
Venetoclax to BCL-2 determines the release of bound BH3-only proteins,
such as BIM, inducing the interaction between these displaced BH3-only
proteins and BAK/BAX with consequent activation of a BAK/BAK complex
(BAK/BAX oligomerization); this activated BAK/BAX complex determines an
increase of the mitochondrial outer membrane permeability (MOMP) with
consequent release of cytochrome c from the mitochondria into the
cytoplasm and activation of the apoptotic process through caspase
activation. In the presence of increased levels of the antiapoptotic
proteins MCL-1, BCL-XL and BCL2-A1, the proapoptotic effect of
Venetoclax in inhibited through a mechanism involving sequestration of
displaced BH3-only proteins, thus preventing the capacity of these
proteins to bind and to activate BAK/BAX and thus blocking the
proapoptotic effect of Venetoclax. |
In
this context, a role particularly relevant is played by MCL-1 as the
main mechanism of VEN resistance. A fundamental study by Bhatt and
co-workers explored the mechanisms of acquired resistance to BH3
mimetic antagonists of BCL-2 and MCL-1 using AML patient-derived
xenograft (PDX) models: these models involved AML cells that have
acquired resistance to VEN following treatment with this drug and AML
cells that were constitutively resistant to VEN.[128]
BH3 profiling studies showed that BH3 mimetic resistance is
characterized by decreased mitochondrial apoptotic priming, as measured
both in primary AML samples and in PDX models, due to alterations at
the level of BCL-2 family proteins, variable from one case to another.[126]
In VEN-resistant AML cells, BCL-2 sequestration of pro-apoptotic
proteins, necessary to confer BCL-2 dependence, was reduced. This
phenomenon was not due to a decreased expression of BCL-2 or BIM after
the acquisition of resistance but to the sequestration of BIM at the
level of MCL-1; an opposite phenomenon is observed in cells resistant
to MCL-1 antagonists, with BIM sequestration at the level of BCL-2.[128]
This finding has considerable implications for therapy based on
combinations of BCL-2 and MCL-1 antagonists since it implies that the
concurrent rather than the sequential administration of these
inhibitors is more effective.[128]
Two initial
studies showed that MCL-1 upregulation renders leukemic cells resistant
to VEN. Thus, Pan et al. provided evidence that a pan-BCLK-2 inhibitor
(-)BI97D6 potently induced apoptosis through intrinsic pathway
activation by disrupting MCL-1/BIM and BCL-2/BAX interactions;
importantly, this pan-BCL-2 inhibitor, as a single agent, effectively
overcame AML cell apoptosis resistance mediated by MCL-1 in AML cells.[129]
The
exploration of VEN-resistant AML cell lines derived through chronic
exposure to VEN showed that the upregulation of MCL-1 and BCL-XL drives
drug resistance; targeting MCL-1 and/or BCL-XL restored the sensitivity of these leukemic cells to VEN.[130]
A consistent number of studies supported the role of MCL-1 in VEN resistance mechanisms.
Ewold and coworkers reported a side-by-side comparison of three different BH3-mimetics targeting BCL-2 or MCL-1, or BCL-XL;
they drove the conclusion that MCL-1 may be a more prevalent
therapeutic target than BCL-2 in AML. Interestingly, MCL-1 BH3-mimetics
induced displacement of the BH3-only protein BIM and BAK, resulting in
BAK-dependent apoptosis; in contrast, VEN-induced cell death was
mediated by BAX rather than BAK.[131] This finding supports distinct non-redundant molecular functions of BCL-2 and MCL-1 in AML cells.
The analysis in vitro
of a large panel of AML cell lines and of primary AML samples
co-cultured with bone marrow mesenchymal stromal cells showed that
inhibition of MCL-1, whose expression is weak compared to that of
BCL-2, induces apoptosis of AML cells and strongly synergizes with VEN.[132]
VU661013
is a selective and potent MCL-1 inhibitor that destabilizes the
association between BIM and MCL-1, induces AML c ells apoptosis, and is
active in VEN-resistant and patients-derived xenografts.[133] Importantly, BH3 profiling of patient samples and ex vivo
drug-sensitivity assays predicted sensitivity to BCL-2 or MCL-1
inhibitors and showed the benefit deriving from the combination of the
two types of inhibitors.[133]
Studies in
pre-clinical models of AMLK supported the double targeting of MCL-1 and
BCL-2. Particularly, the analysis of primary AML samples, including
those with poor risk genotypes, showed the efficacy of the
contemporaneous MCL-1 and BCL-2 targeting in the induction of leukemic
cell apoptosis.[134] Furthermore, co-targeting of
MCL-1 and BCL-2 was more effective against the leukemic compartment
compared to normal hematopoietic stem/progenitor cells, thus supporting
a therapeutic window of activity and a tolerable safety profile at the
level of the hematopoietic system.[134] Importantly,
BCL-2 and MCL-1 co-targeting prolonged animal survival in xenograft
models of AML and suppressed patient-derived leukemia but not normal
hematopoietic cells in the bone marrow of engrafted animals.[134]
A
recent study showed that co-targeting of BCL-2 and MCL-1 was highly
effective in inhibiting leukemic stem cells derived from AMLs resistant
to VEN or relapsed after VEN-based therapy, irrespective of genetic
alterations and cytogenetic alterations.[127]
Interestingly, enhanced antileukemia activity was also observed in a
PDX model of monocytic leukemia, a leukemia subtype known to be
resistant to VEN-based therapy.[135]
Importantly,
a recent study provided evidence that the BCL-2 and MCL-1 co-targeting
was effective not only against VEN-resistant AML cells but also against
chemotherapy-resistant cells. Accordingly, S63845, an MCL-1 inhibitor,
in combination with VEN, effectively inhibits AML cells that have
acquired resistance to cytarabine, thus supporting an evaluation of
this drug combination in relapsed/refractory AML patients.[136]
Proteasome
inhibition may represent a new strategy to antagonize MCL-1 activity in
cancer cells via transactivation of the MCL-1 antagonist NOXA.[137] Interestingly, proteasome inhibitors strongly synergize with VEN in inducing apoptosis of cancer cells.[137] However, this synergistic combination was not yet evaluated in preclinical models of AML.
The
increased dependency on MCL-1 observed constitutively in some AMLs or
following VEN treatment may be related to activation of the MAPK
signaling pathway, which induces stabilization of MCL-1 protein and
prevents its degradation.[138-139] RAS-MAPK
activation is a major mechanism of acquired VEN resistance, mediated by
MCL-1 upmodulation and that can be abrogated by MCL-1 inhibition; rapid
clonal selection of RAS-mutated clones was observed in some AML
patients treated with VEN-containing regimens and represented an
important mechanism underlying development of VEN resistance.[140]
RAS pathway mutations, such as those occurring at the level of KRAS and PTPN11 genes, induce VEN resistance to AML cells, related to a mechanism involving MCL-1 and BCL-XL upmodulation; these AML subtypes are sensitive to MCL-1 inhibitors.[138] Studies in AML cell lines have shown that VEN resistance was reproduced through transduction of G12D KRAS or A72D PTPN11 mutants; G12D KRAS-transduced cells displayed decreased BCL-2 and BAX levels, associated with increased MCL-1 and BCL2-A1 levels.[138] Only MCL-1 inhibitors, but not BCL-2 or BCL-XL inhibitors, could reduce the viability of these leukemic cells.[138] The combination of VEN+MCL-1 inhibitors showed synergy in inducing apoptosis of leukemic cells.[138]
RAS
pathway activation, observed in RAS- and PTPN11-mutated cases, induces
the activation of multiple sources of energy for cell survival,
including fatty acid and amino acid metabolism, glycolysis and
upregulation of OXPHOS.[141] Furthermore, AMLs harboring a PTPN11 mutation frequently display a monocytic differentiation phenotype,[142] usually associated with dependency on MCL-1 and thus scarcely sensitive to VEN-mediated BCL-2 inhibition.[143]
In
line with these findings, inhibition of the MAPK signaling pathway
using MEKK/MEK2 inhibitors synergizes with VEN to induce apoptosis of
AML cells.[144] Furthermore, this drug combination
induced downregulation of MCL-1 levels and disrupted the binding of BIM
to both MCL-1 and BCL-2, thus releasing BIM that was able to initiate
the apoptotic process.[144] The observations
provided a rationale for the combinatorial blockade of MEK and BCL-2
pathways in AML subsets characterized by KRAS, NRAS, PTPN11, and FLT3-ITD mutations.
Ongoing
phase I clinical trials are evaluating different MCL-1 inhibitor-based
treatments for AML patients, including the combination therapies with
VEN (NCT03672695, NCT02979366, and NCT04629443).
Interestingly, a
recent phase It study showed that VEN with low-dose Navitoclax was well
tolerated and had promising efficacy in patients with
relapsed/refractory acute lymphoblastic leukemia or lymphoblastic
lymphoma.[145] Therefore, an ongoing clinical study
is evaluating the combination of VEN, Navitoclax, and Decitabine in
relapsed/refractory AML patients previously treated with VEN
(NCT05222984).
Conclusions
The
advent of VEN as a potent BCL-2 inhibitor has transformed the treatment
of AML, particularly for elderly patients. Multiple VEN-based
combination therapies have been developed based on understanding the
mechanisms underlying VEN responsiveness and resistance (primary and
adaptive). Numerous studies carried out in the last years have shown
that de novo-treated AML patients respond to these treatments better than refractory/relapsing patients.
Retrospective
studies involving molecular characterization of treated patients have
shown that the most responding patients are represented by those with NPM1, IDH1-IDH2, and TET2 mutations; RUNX1mutated
patients, both in frontline and in relapsing/refractory status, seem to
be more responsive to VEN-based regimens than to intensive
chemotherapy; FLT3-mutant
AMLs shows a reduced sensitivity to VEN+HMA or VEN+LDAC regimens, but
could be more responsive to “triplet” regimens based on VEN+FLT3
inhibitors+HMA (or LDAC); AML patients harboring TP53, RAS, or PTPN11 mutations, monocytic AML, secondary AML and AML cases pre-treated with HMAs show reduced sensitivity to VEN-based therapies.
The ongoing development of “triplet”
therapies based on the administration of VEN+HMA 8 or LDAC) + a drug
targeting a specific molecular alteration or a signaling pathway could
significantly improve the rates and the duration of the clinical
responses. However, the development of these “triplet” therapies could
be hampered in some instances by the occurrence of not tolerable
toxicities.
Another important development will consist in the
association of VEN with another BH3 targeting drug, such as MCL-1 or
BCL-XL inhibitor, to extend the number of responding patients and
bypass VEN primary and adaptive resistance.
References
- Kale J, Osterlund EJ, Andrews DW. BCL-2 family
proteins: changing partners in the dance towards death. Cell Death
Differ 2018; 25: 65-80. https://doi.org/10.1038/cdd.2017.186 PMid:29149100 PMCid:PMC5729540
- Soners
AJ, Lavenon JD, Boghaert ER, et al. ABT-199, a potent and selective
BCL-2 inhibitor, achieves antitumor activity while sparing platelets.
Nature Med 2013; 19: 202-208. https://doi.org/10.1038/nm.3048 PMid:23291630
- Pan
R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J,
DeAngelo DJ, Debose L, et al. Selective BCL-2 inhibition by ABT-199
causes on-target cell death in acute myeloid leukemia. Cancer Discov
2014; 4: 362-375. https://doi.org/10.1158/2159-8290.CD-13-0609 PMid:24346116 PMCid:PMC3975047
- Laagdinou
ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM,
Pei S, Grose V, O'Dwyer KM, et al. BCL-2 inhibition targets oxidative
phosphorylation and selectively eradicates quiescent humabn leukemic
stem cells. Cell Stem Cell 2013; 12: 329-341. https://doi.org/10.1016/j.stem.2012.12.013 PMid:23333149 PMCid:PMC3595363
- DiNardo
CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, Arellano M,
Frattini MG, Kantarjian H, Popovic R, et al. Safety and preliminary
efficacy of venetoclax with decitabine or azacytidine in elderly
patients with previously untreated acute myeloid leukemia: a
non-randomized, open-label, phase 1b study. Lancet Oncol 2018; 19(2):
216-228. https://doi.org/10.1016/S1470-2045(18)30010-X
- DiNardo
CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O,
Konopleva M, Wei AH, Kantarjian HM, et al. Venetoclax combined with
decitabine or azacytidine in treatment-naïve, elderly patients with
acute myeloid leukemia. Blood 2019; 133(1): 7-17. https://doi.org/10.1182/blood-2018-08-868752 PMid:30361262 PMCid:PMC6318429
- Wei
AH, Strickland SA, Hou JZ, Fiedler W, Lin TL, Walter RB, Enjeti A,
Tiong IS, Savona M, Lee S, et al. Venetoclax comvined with low-dose
cytarabine for previously untreated patients with acute myeloid
leukemia: results from a phase Ib/II study. J Clin Oncol 2019; 37(15):
1277-1284. https://doi.org/10.1200/JCO.18.01600 PMid:30892988 PMCid:PMC6524989
- DiNardo
CD, Jonas BA, Poullarkat V, Thirman MJ, Garcia JS, Wei AH, Konopleva M,
Dohner H, Letai A, Fenaux P, et al. Azacitidine and venetoclax in
previously untreated acute myeloid leukemia. N Engl J Med 2020; 383(7):
617-629. https://doi.org/10.1056/NEJMoa2012971 PMid:32786187
- Wei
AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K,Kim I,
Stevens DA, Fiedler W, Pagoni M, et al. Venetoclax plus LDAC for newly
diagnosed AML ineligible for intensive chemotherapy: a phase 3
randomized placebo-controlled trial. Blood 2020; 135(24): 2137-2145. https://doi.org/10.1182/blood.2020004856 PMid:32219442 PMCid:PMC7290090
- Pratz
KW, Jonas BA, Pullarkat V, Recher C, Schuh AC, Thirman MJ, Garcia JS,
DiNardo CD, Vorobyev V, Fracchiolla NS, et al. Measurable residual
disease response and prognosis in treatment-naïve acute myeloid
leukemia with venetoclax and azacytidine. J Clin Oncol 2022; 40(8):
855-865. https://doi.org/10.1200/JCO.21.01546 PMid:34910556
- Winters
AC, Gutman JA, Purev E, Nakic M, Tobin J, Chase S, Kaiser J, Lile L,
Boggs C, Halsema K, et al. Real-world experience of venetoclax with
azacytidine for untreated patients with acute myeloid leukemia. Blood
Adv 2019; 3(20): 2911-2920. https://doi.org/10.1182/bloodadvances.2019000243 PMid:31648312 PMCid:PMC6849960
- Pollyea
DA, Pratz K, Letai A, Jonas BA, Wei AH, Pullarkat V, Konopleva M,
Thirman MJ, Arellano M,Becker PS, et al. Venetoclax with azacitidine or
decitabine in patients with newly diagnosed acute myeloid leukemia:
long term follow-up from a phase 1b study. Am J Hematol 2021; 96:
208-217. https://doi.org/10.1002/ajh.26039 PMid:33119898
- DiNardo
CD, Maiti A, Rausch CR, Pemmaraju N, Naqvi K, Daver NG, Kadia TM,
Borthakur G, Ohanina M, Alvarado Y, et al. 10-day decitabine with
venetoclax for newly diagnosed intensive chemotherapy ineligible, and
relapsed or refractory acute myeloid leukemia: a single-centre, phase 2
trial. Lancet Hematol 2020; 7(10): e724-e736. https://doi.org/10.1016/S2352-3026(20)30210-6
- DiNardo
CD, Rausch CR, Pemmaraju N, Garcia-Manero G, Ohanian M, Daver N, Issa
GC, Borthakur G, Ravandi F, Alvarad Y, et al. Phase II trial of ten-day
decitabine plus venetoclax (DEC10-VEN) in acute myeloid leukemia:
updated outcomes in genomic subgroups. Blood 2021.
- Maiti
A, Qiao W, Sasaki K, Ravandi F, Kadia TM, Jabbour EJ, Daver NG,
Borthakur EJ, Garcia-Manero G, Pierce SA, et al. Venetoclax with
decitabine vs intensive chemotherapy in acute myeloid leukemia: a
propensity score matched analysis stratified by risk of
treatment-related mortality. Am J Hematol 2021; 96: 282-291. https://doi.org/10.1002/ajh.26061 PMid:33264443 PMCid:PMC8128145
- Kadia
TM, Reville PK, Wang X, Rausch CR, Borthakur G, Pemmaraju N, Daver NG,
DiNardo CD, Sasaki K, Issa GC, et al. Phase II study of venetoclax
added to cladribine plus low-dose cytarabine alternating with
5-azacitidine in older patients with newly diagnosed acute myeloid
leukemia. J Clin Oncol 2022; in press. https://doi.org/10.1200/JCO.21.02823 PMid:35704787
- Daver
N, Konopleva M, Maiti A, Kadia TM, DiNardo CD, Loghavi S, Pemmaraju N,
Jabbour EJ, Montalban-Bravo G, Tang G, et al. Phase I/II study of
azacytidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in
patients (pts) with newly diagnosed older/unfit or high-risk acute
myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood 2021;
abst 616. https://doi.org/10.1182/blood-2021-153638
- Chua
CC, Roberts AW, Reynolds J, Fong CY, Ting SB, Salmon JM, MacRaild S,
Ivey A, Tiong IS, Fleming S, et al. Chemotherapy and venetoclax in
elderly acute myeloid leukemia trial (CAVEAT): a phase Ib
dose-escalation study of venetoclax combined with modified intensive
chemotherapy. J Clin Oncol 2020; 38(30): 3506-3517. https://doi.org/10.1200/JCO.20.00572 PMid:32687450
- DiNardo
C, Lachowiez CA, Takahashi K, Loghavi S, Xiao L, Kadia T, Daver N,
Adeoti M, Short NJ, Sasaki K, et al. Venetoclax combined with FLAG-IDA
induction and consolidation in newly diagnosed and relapsed or
refractory acute myeloid leukemia. J Clin Oncol 2021; 39(25):
2768-2778. https://doi.org/10.1200/JCO.20.03736 PMid:34043428 PMCid:PMC8407653
- DiNardo
CD, Lachowiez CA, Takahashi K, Loghavi S, Kadia T, Daver N, Xiao L,
Adeoti M, Short NJ, Sasaki K, et al. Venetoclax combined with FLAG-IDA
induction and consolidation in newly diagnosed acute myeloid leukemia.
Am J Hematol 2022; 97(8): 1035-1043. https://doi.org/10.1002/ajh.26601 PMid:35583199
- Lachowiezv
CA, Reville PK, Kantarjian H, Jabbour E, Bothakur G, Daver N, Loghavi
S, Furudate K, Xiao L, Pierce S, et al. Venetoclax combined with
induction chemotherapy in patients with newly diagnosed acute myeloid
leukemia: a post-hoc, propensity score-matched, cohort study. Lancet
Hematol 2022; 9(5): e350-e360. https://doi.org/10.1016/S2352-3026(22)00076-X
- Stone
RM, De Angelo DJ, Galinsky I. Phasi I trial of escalating doses of the
Bcl-2 inhibitor venetoclax in combination with daunorubicin/cytarabine
induction and high dose cytarabine consolidation in previously
untreated adults with acute myeloid leukemia (AML). Blood 2019; 134:
3908-3908. https://doi.org/10.1182/blood-2019-124966
- Stone
RM, De Angelo DJ, Letai AG. Maximal tolerated dose of the BCL-2
inhibitor venetoclax in combination with daunorubicin/cytarabine
induction in previously untreated adults with acute myeloid leukemia
(AML). Blood 2020; 136: 40-41. https://doi.org/10.1182/blood-2020-140646
- Wang
H, Mao L, Yang M, Qian P, Lu H, Tong H. Ventoclax plus 7+3 daunorubicin
and cytarabine therapy as first-line treatment for adults with acute
myeloid leukemia: a multicentre, single-arm, phase 2 trial. Lancet
Hematol 2022; 9(6): E415-E424. https://doi.org/10.1016/S2352-3026(22)00106-5
- Kim
K, Kantarjian H, Borthakur G, Takahashi K, Short NJ, DiNardo CD,
Jabbour EJ, Chien K, Daver N, Pemmaraju N, et al. A phase II study of
CPX-351 plus venetoclax in patients with relapsed/refractory (R/R) or
newly diagnosed acute myeloid leukemia (AML). Blood 2021; 138 (suppl
1): 1275. https://doi.org/10.1182/blood-2021-154148
- Uy
GL, Pullarkat VA, Baratam P, Stuart RK, Walter RB, Winer ES.
Lowerintensity CPX-351 + venetoclax for patients with newly diagnosed
AML who are unfit for intensive chemotherapy. J Clin Oncol 2022; 40,
suppl. 16: 7031. https://doi.org/10.1200/JCO.2022.40.16_suppl.7031
- Konopleva
M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E,
Hamed Salem A, Zhu M, Ricker JL, et al. Efficacy and biological
correlates of response in a phase II study of venetoclax monotherapy in
patients with acute myelogenous leukemia. Cancer Discover 2016; 6(10):
1106-1117. https://doi.org/10.1158/2159-8290.CD-16-0313 PMid:27520294 PMCid:PMC5436271
- DiNardo
CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, Daver N, Covert
W, Marx KR, Mace M, et al. Clinical experience with the BCL2-inhibitor
venetoclax in combination therapy for relapsed and refractory acute
myeloid leukemia and related myeloid malignancies. Am J Hematol 2018;
93(3): 401/407. https://doi.org/10.1002/ajh.25000 PMid:29218851
- Aldoss
I, Yang D, Aribi A, Ali H, Sandhu K, Malki M, Mei M, Salhotra A, Khaled
S, Nakamura R, et al. Efficacy of the combination of venetoclax and
hypomethylating agents in relapsed/refractory acute myeloid leukemia.
Haematologica 2018; 103: e405. https://doi.org/10.3324/haematol.2018.188094 PMid:29545346 PMCid:PMC6119155
- Stahl
M, Menghrajani K, Derkach A, Chan A, Xia W, Glass J, King AC, Daniyan
AF, Famulare C, Cuello Bm, et al. Clinical and molecular predictors of
response and survival following venetoclax therapy in
relapsed/refractory AML. Blood Adv 2021; 5(5): 1552-1564. https://doi.org/10.1182/bloodadvances.2020003734 PMid:33687434 PMCid:PMC7948282
- Labrador
J, Saiz-Rodriguez M, de Miguel D, de Laiglesia A, Rodriguez-Medina C,
Vidriales MB, Perez-Encina M, Sanchez-Sanchez MJ, Cuello R, Rodan-Perez
A, et al. Use of venetoclax in patients with relapsed or refractory
acute myeloid leukemia: the PETHEMA registry experience. Cancers 2022;
14(7): 1734. https://doi.org/10.3390/cancers14071734 PMid:35406512 PMCid:PMC8997036
- Feld
J, Tremblay D, Dougherty M, Czaplinska T, Sanchez G, Brady C,
Kremyanskaya M, Bar-Natan M, Keyzner A, Marcellino BK, et al. Safety
and efficacy: clinical experience of venetoclax in combination with
hypomethylating agents in both newly diagnosed and relapsed/refractory
advanced myeloid malignancies. Hemasphere 2021; 5(4): e549. https://doi.org/10.1097/HS9.0000000000000549 PMid:33718803 PMCid:PMC7951133
- Wang
YW, Tsai CH, Lin CC, Tien FM, Chen YW, Lin HY, Yao M, Lin YC, Lin CT,
Cheng CL, et al. Cytogenetics and mutations could predict outcome in
relapsed and refractory acute myeloid leukemia patients receiving BCL-2
inhibitor venetoclax. Ann Hematol 2020; 99(3): 501-511. https://doi.org/10.1007/s00277-020-03911-z PMid:31965269
- Maiti
A, DiNardo CD, Qiao W, Kadia TM, Jabbour EJ, Rausch CR, Daver NG, Short
NJ, Borthakur G, Pemmaraju N, et al. Ten-day decitabine with venetoclax
versus intensive chemotherapy in relapsed or refractory acute myeloid
leukemia: a propensity score-matched analysis. Cancer 2021; 127(22):
4213-4220. https://doi.org/10.1002/cncr.33814 PMid:34343352
- Wolach
O, Frisch A, Shargian L, Yeshurun M, Apel A, Vainstein V, Moshe Y,
Shimony S, Amit O, Bar-On Y, et al. Venetoclax in combination with
FLAG-IDA-based protocol for patients with acute myeloid leukemia: a
real-world analysis. Ann Hematol 2022; in press. https://doi.org/10.1007/s00277-022-04883-y PMid:35732976
- Shahswar
R, Beutel G, Klement P, Rehberg A, Gabdoulline R, Koenecke C, Markel D,
Eggers H, Eder M, Stadler M, et al. FLAG-IDA salvage chemotherapy
combined with a seven-day course of venetoclax (FLAVIDA) in patients
with relapsed/refractory acute leukemia. Brit J Haematol 2020; 188:
e1-e30A https://doi.org/10.1111/bjh.16268 PMid:31681986 PMCid:PMC7116720
- Ravandi
F, Abuasab T, Alvarado Valero Y, Issa GC, Islam R, Short NJ, Yilmaz M,
Jain N, Masarova L, Kornblau SM, et al. Phase 2 study of ASTX727
(cedazuridine/decitabine) plus venetoclax (ven) in patients with
relapsed/refractory acute myeloid leukemia (AML) or previously
untreated, elderly patients (pts) unfit for chemotherapy. J Clin Oncol
2022; 40: 16 suppl.1, abst. 7037. https://doi.org/10.1200/JCO.2022.40.16_suppl.7037
- Montesinos
P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, Heuser M, Calado
RT, Schuh AC, Yeh SP, et al. Ivosidenib and azacytidine in IDH1-mutated
acute myeloid leukemia. N Engl J Med 2022; 386: 1519-1531. https://doi.org/10.1056/NEJMoa2117344 PMid:35443108
- Lachowiez
CA, Suen Garcia J, Borthakur G, Loghavi S, Zeng Z, Tippett GD, Kadia
TM, Masarova L, Yilmaz M, Maiti A, et al. A phase Ib/II study of
ivosidenib with venetoclax +/- azacytidine in IDH1-mutated hematologic
malignancies. J Clin Oncol 2022; 40(16): suppl. Abst. 7018. https://doi.org/10.1200/JCO.2022.40.16_suppl.7018
- Venugopal
S, Maiti A, DiNardo CD, Loghavi S, Daver NG, Kadia TM, Rausch CR,
Alvarado Y, Ohanian M, Sasaki K, et al. Decitabine and venetoclax for
IDH1/2-mutated acute myeloid leukemia. Am J Hematol 2021; 96(5):
E154-E157. https://doi.org/10.1002/ajh.26122 PMid:33580980 PMCid:PMC8237705
- Venugopal
S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, Short NJ,
Ohanian M, Masarova L, Issa G, et al. Efficacy and safety of enasidenib
and azacytidine combination in patients with IDH2 mutated acute myeloid
leukemia and not eligible for intensive chemotherapy. Blood Cancer J
2022; 12:10. https://doi.org/10.1038/s41408-021-00604-2 PMid:35078972 PMCid:PMC8789767
- Chua
CC, Hammond D, Kent A, Tiong IS, Konopleva MY, Pollyea DA, DiNardo CD,
Wei AH. Treatment-free remission after ceasing venetoclax-based therapy
in patients with acute myeloid leukemia. Blood Adv 2022; in press. https://doi.org/10.1182/bloodadvances.2022007083 PMid:35511730 PMCid:PMC9278306
- Pollyea
DA, Winters A, McMahon C, Schwartz M, Jordan CT, Rabinovitch R, Abbott
D, Smith CA and Gutman JA. Venetoclax and azacytidine followed by
allogeneic transplant results in excellent outcomes and may improve
outcomes versus maintenance therapy among newly diagnosed AML patients
older than 60. Bone Marrow Transpl 2022; 57: 160-166. https://doi.org/10.1038/s41409-021-01476-7 PMid:34645926
- Kent
A, Pollyea DA. Winters A, Jordan CT, Smith C, Gutman JA. Venetoclax is
safe and tolerable as post-transplant maintenance therapy for AML
patients at high risk for relapse. Blood 2020; 134: 2410. https://doi.org/10.1182/blood-2020-138832
- Bazinet
A, Kantarjian H, Borthakur G, Yilmaz M, Bose P, Jabbour EJ, Alvarado Y,
Chien KS, Pemmaraju N, Takahashi K, et al. A phase II study of
5-azacitidine (AZA) and venetoclax as maintenance therapy in patients
with acute myeloid leukemia (AML) in remission. Blood 2021; 138:
supll.1: 2326. https://doi.org/10.1182/blood-2021-150573
- Bazinet
A, Kantarjian H, Borthakur G, Yilmaz M, Bose P, Jabbour EJ, Alvarado Y,
Chien KS, Pemmaraju N, Takahashi K, et al. Phase 2 study of azacytidine
(AZA) and venetoclax (VEN) as maintenance therapy for acute myeloid
leukemia (AML) patients in remission. J Clin Oncol 2022; 40(16). Suppl.
Abst. E19018. https://doi.org/10.1200/JCO.2022.40.16_suppl.e19018
- Lachowiez
CA, Loghavi S, Kadia TM, Daver N, Borthakur G, Pemmaraju N, Naqvi K,
Alardo V, Yilmaz M, Short N, et al. Outcomes of older patients with
NPM1-mutated AML: current treatment and the promise of venetoclax-based
regimens. Blood Adv 2020; 4(7): 1311-1320. https://doi.org/10.1182/bloodadvances.2019001267 PMid:32251497 PMCid:PMC7160266
- Tiang
IS, Dillon R, Ivey A, The TC, Nguyen P, Cummings N, Taussig DC, Latif
AL, Potter NE, Runglall M, et al. Brit J Haematol 2021; 192(6):
1026-1030. https://doi.org/10.1111/bjh.16722 PMid:32458446 PMCid:PMC8048658
- Begenberger
JM, Kornblau SM, Piercall WE, Lena R, Cow D, Shi CX, Mantel J, Ahmann
G, Gonzalez JM, Choudhary A, et al. BCL-2 family proteins and
5-azacytidine-sensitizing targets and determinants of response in
myeloid malignancies. Leukemia 2014; 28: 1657-1665. https://doi.org/10.1038/leu.2014.44 PMid:24451410 PMCid:PMC4131248
- Jones
CL, Stevens BM, D'Alessandro A, Reisz Á, Culp-Hill R, Nemkov T, Pei S,
Khan N, Adane B, Ye H, et al. Inhibition of amino acid metabolism
selectively targets human leukemia stem cells. Cancer Cell 2018; 34:
724-740.e4. https://doi.org/10.1016/j.ccell.2018.10.005 PMid:30423294 PMCid:PMC6280965
- Pollyea
DA, AStevens BM, Jones CL, Winters A, Pei S, Minhajuddin M,
D'Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. Venetoclax
with azacytidine disrupts energy metabolism and targets leukemia stem
cells in patients with acute myeloid leukemia. Nat Med 2018; 24:
1859-1866. https://doi.org/10.1038/s41591-018-0233-1 PMid:30420752 PMCid:PMC7001730
- Stevens
BM, Jones CL, Pollyea DA, Culp-Hill R, D'Alessandro A, Winters A, Krug
A, Abbott D, Goosman M, Pei S, et al. Fatty acid metabolism underlies
venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer
2020; 1(12): 1176-1187. https://doi.org/10.1038/s43018-020-00126-z PMid:33884374 PMCid:PMC8054994
- Jones
CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, Gehrke S,
Gamboni F, Krug A, Winters A, et al. Nicotinamide metabolism mediates
resistance to venetoclax in relapsed acute myeloid leukemia stem cells.
Cell Stem Cell 2020; 27(5): 748-764.e4. https://doi.org/10.1016/j.stem.2020.07.021 PMid:32822582 PMCid:PMC7655603
- Bisaillon
R, Moison C, Thiollier C, Krosl J, Bordeleau ME, Lehnertz B, Lavallée
JP, MacRae C, Boucher G, et al. Genetic characterization of ABT-199
sensitivity in human AML. Leukemia 2020; 34: 63-74. https://doi.org/10.1038/s41375-019-0485-x PMid:31300747
- Kontro
M, Kumar A, Majumder MM, Eldfors S, Parson A, Pemovska T, et al. HOX
gene expression predicts response to BCL-2 inhibition in acute myeloid
leukemia. Leukemia 2017; 31: 301-319. https://doi.org/10.1038/leu.2016.222 PMid:27499136
- Chesnokov
MS, Borhani S, Halasi M, Arbieva Z, Zhan I, Gartel AL.
FOXM1-AKT-positive regulation loop provides venetoclax resistance in
AML. Front Oncol 2021; 11: 696532. https://doi.org/10.3389/fonc.2021.696532 PMid:34381718 PMCid:PMC8350342
- Wu
HC, Rérolle D, Berthier C, Hleihel R, Sakamoto T, Quentin S, Benhenda
S, Morganti C, Wu C, Conte L, et al. Actinomycin D targets NPM1c-primed
mitochondria to restore PML-driven senescence in AML therapy. Cancer
Discovery 2021; 11(12): 3198-3213. https://doi.org/10.1158/2159-8290.CD-21-0177 PMid:34301789 PMCid:PMC7612574
- Ranieri
R, Piangiani G, Perriello VM, Marra A, Cardinali V, Pierangeli S,
Milano F, Gianfriddo I, Brunetti L, Martelli MP, Falini B. Current
status and future perspectives in targeted therapies of NPM1-mutated
AML. Leukemia 2022; in press. https://doi.org/10.1038/s41375-022-01666-2 PMid:36008542 PMCid:PMC9522592
- Fisher
MA, Friedlander SY, Arrate MP, Chang H, Gorska AE, Fuller RD, Ramsey
HE, Kaskyap T, Argueta C, Debler S, et al. Venetoclax response is
enhanced by selective inhibitor of nuclear export compounds in
hematological malignancies. Blood Adv 2020; 4(3): 586-598. https://doi.org/10.1182/bloodadvances.2019000359 PMid:32045477 PMCid:PMC7013257
- Pangiani
G, Gagliardi A, Mezzaroma F, Rocchio F, Tini V, Bigema B, Sportolketti
P, Caruso S, Marra A, Peruzzi S, et al. Prolonged XPO1 inhibition is
essential for antileukemic activity in NPM1-mutated AML. Blood Adv
2022; in press. https://doi.org/10.1182/bloodadvances.2022007563 PMid:36037515
- Klossowski
S, Miao H, Kempinska K, Wu T, Purohit T, Kim E, Linhares BM, Cghen D,
Jih G, Perkey E, et al. Menin inhibitor MI-3454 induces remission in
MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest
2020; 130(2): 981-997. https://doi.org/10.1172/JCI129126 PMid:31855575 PMCid:PMC6994154
- Uckelmann
HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, Krivstov AV,
Rucker FG, Dohner K, McGeehan GM, et al. Therapeutic targeting of
preleukemia cells in a mouse model of NPM1 mutant acute myeloid
leukemia. Science 2020; 367(6477): 586-590. https://doi.org/10.1126/science.aax5863 PMid:32001657 PMCid:PMC7754791
- Carter
BZ, Tao W, Mak PY, Ostermann LB, Mak D, McGeehan G, et al. Menin
inhibition decreases Bcl-2 and synergizes with venetoclax in
NPM1/FLT3-mutated AML. Blood 2021; 138: 1637-1641. https://doi.org/10.1182/blood.2021011917 PMid:34232981
- Fiskus
W, Boettcher S, Daver N, Mill CP, Sasaki K, et al. Effective menin
inhibitor-based combinations against AML with MLL rearrangement or NPM1
mutation (NPM1c). Blood Cancer J 2022; 12: 5. https://doi.org/10.1038/s41408-021-00603-3 PMid:35017466 PMCid:PMC8752621
- Venugopal
S, Maiti A, DiNardo CA, Loghavi S, Daver DG, Kadia TM, Rausch CR,
Alvarado Y, Ohanion M, Sasaki K, et al. Decitabine and venetoclax for
IDH1/2-mutated acute myeloid leukemia. Am J Hematol 2021; 96(5):
E154-E157. https://doi.org/10.1002/ajh.26122 PMid:33580980 PMCid:PMC8237705
- Hammond
D, Loghavi S, Konopleva M, Response patterns and MRD by flow cytometry
and NGS in patients with mutant-IDH acute myeloid leukemia treated with
venetoclax in combination with hypomethylating agents. Blood 2020;
136(suppl. 1): 35-36. https://doi.org/10.1182/blood-2020-141380
- Pollyea
DA, DiNardo CA, Arellano ML, Pigneux A, Fiedler W, Konopleva M,
Rizzieri DA, Smith D, Shinagawa A, Memoli R, et al. Impact of
venetoclax and azacytidine in treatment-naïve patients with acute
myeloid leukemia and IDH 1-2 mutations. Cancer Res 2022; 28: 2753-2761.
https://doi.org/10.1158/1078-0432.CCR-21-3467 PMid:35046058
- Montesinos
P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, Heuser M, Calado
RT, Schuh AC, Yeh SP, et al. Ivosidenib and zacitidine in IDH1-mutated
acute myeloid leukemia. N Engl J Med 2022; https://doi.org/10.1056/NEJMoa2117344 PMid:35443108
- Botton
S, Chol S, Marchione DM, et al. Molecular characterization of clinical
response in patients with newly diagnosed AML treated with ivosidenib +
azacytidine compared to placebo + azacytidine. J Clin Oncol 2022;
40(suppl. 16): 7019. https://doi.org/10.1200/JCO.2022.40.16_suppl.7019
- DiNardo
CD, Schuh AC, Stein EM, Montesinos P, Wei AH, de Botton S, Zeidan AM,
Fathi AT, Kantarjian HM, Bennett JM, et al. Enasidenib plus azacytidine
versus azacytidine alone in patients with newly diagnosed, mutant-IDH2
acute myeloid leukemia (AG221-AML-005): a single-arm, phase 1b and
randomized phase 2 trial. Lancet Oncol 2021; 22(11): 1597-1608. https://doi.org/10.1016/S1470-2045(21)00494-0
- Lachowiez
CA, Bethakur G, Loghavi S, Zeng Z, Kadia TM, Masarova L, Takahashi K,
Tippett GD, Smith S, Garcia JS, et al. A phase Ib/II study of
ivosidenib with venetoclax +/- azacytidine in IDH1-mutated myeloid
malignancies. J Clin Oncol 2021; 39(suppl. 15): 7012. https://doi.org/10.1200/JCO.2021.39.15_suppl.7012
- Lachowiez
AC, Garcia JS, Borthakur G, Loghavi S, Zeng Z, Tippett GD, Kadia TM,
Masorava L, Yilmaz M, Maiti A, et al. A phase Ib/II study of ivosidenib
with venetoclax +/- azacytidine in IDH1-mutated hematologic
malignancies. J Clin Oncol 2022; 40(suppl. 16):7018-7018. https://doi.org/10.1200/JCO.2022.40.16_suppl.7018
- Venugopal
S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, Short NJ,
Ohanian M, Masarova L, Issa G, et al. Efficacy and safety of enasidenib
and azacytidine combination in patients with IDH2 mutated acute myeloid
leukemia and not eligible for intensive chemotherapy. Blood Cancer J
2022; 12(1): 10. https://doi.org/10.1038/s41408-021-00604-2 PMid:35078972 PMCid:PMC8789767
- Chan
SM, Cameron C, Chatelin S, Gupta V, More D, Minden MD, Murphy T,
Schimman AD, Schuh AC, Sibai H, et al. Enasidenib in combination with
venetoclax in IDH2-mutated myeloid malignancies: preliminary results of
the phase I/II Enaven-AML trial. Blood 2021; 138(suppl.1): 1263. https://doi.org/10.1182/blood-2021-153660
- Aldoss
I, Yand D, Pillai R, Sanchez JF, Mei M, Aribi A, Ali H, Sandhu K, Al
Malki M, Salhotra A, et al. Association of leukemia genetics with
response to venetoclax and hypomethylating agents in
relapsed/refractory acute myeloid leukemia. Am J Hematol 2019; 94:
E253-E255. https://doi.org/10.1002/ajh.25567 PMCid:PMC8855495
- Chan
SM, Thomas D, Coprces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F,
Medeiros BC, Tyvoll DA, Majeti R. Isocitrate dehydrogenase 1 and 2
mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med
2015; 21: 178-184. https://doi.org/10.1038/nm.3788 PMid:25599133 PMCid:PMC4406275
- Konopleva
M, Thirman M, Pratz KW, Letai AG, Recher C, Pullarkat VA, et al.
Results of venetoclax and azacytidine combination in chemotherapy
ineligible untreated patients with acute myeloid leukemia with FLT3
mutations. Blood 2020; 136: 8-10. https://doi.org/10.1182/blood-2020-134100
- Konopleva
M, Thirman MJ, Pratz KW, Garcia JS, Recher C, Pullarkat V, Kantarjian
HM, DiNardo CD, Dail M, Duan Y, et al. Impact of FLT3 mutation on
outcomes after venetoclax and azacytidine for patients with
treatment-naïve acute myeloid leukemia. Clin Cancer Res 2022; in press.
https://doi.org/10.1158/1078-0432.CCR-21-3405 PMid:35063965
- DiNardo
CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, Thijson R,
Pomilio G, Ivey A, Salmon JM, et al. Molecular patterns of response and
treatment failure after frontline venetoclax in older patients with
AML. Blood 2020; 135(11): 791-803. https://doi.org/10.1182/blood.2019003988 PMid:31932844 PMCid:PMC7068032
- Mali
RS, Zhang Q, Di Filippis RA, Cavazos A, Kuruvilla A, Roman J, Mody V,
Choo E, Dail M, Shah NP, Konopleva M, et al. Venetoclax combines
synergistically with FLT3 inhibition to effectively target leukemic
cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021;
104(6): 1034-1046. https://doi.org/10.3324/haematol.2019.244020 PMid:32414851 PMCid:PMC8017817
- Brinton
LT, Zhang P, Williams K, Canfield D, Orwick S, Sher S. Synergistic
effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J
Hematol Oncol 2020; 13: 139. https://doi.org/10.1186/s13045-020-00973-4 PMid:33076970 PMCid:PMC7574303
- Zhu
R, Li L, Nguyen B, Seo J, Wu M, Seale T. FLT3 tyrosine kinase
inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute
leukemia cells through BIM activation. Signal Transduct Target Ther
2021; 6: 186. https://doi.org/10.1038/s41392-021-00578-4 PMid:34024909 PMCid:PMC8141515
- Altman
JK, Daver N, Maly J. AML-162: efficacy and safety of venetoclax in
combination with gilteritinib for relapsed/refractory FLT3-mutated
acute myeloid leukemia: updated analyses of a phase 1b study. Clin
Lymphoma Myeloma Leuk 2021; 21: 5285. https://doi.org/10.1016/S2152-2650(21)01684-0
- Dover
N, Perl AE, Maly J, Levis M, Ritchie E, Litzow M, McCloskey J, Smith
CC, Schiller G, Bradley T, et al. Venetoclax plus geltiritinib for
FLT3-mutated relapsed/refractory acute myeloid leukemia. J Clin Oncol
2022; in press. https://doi.org/10.1200/JCO.22.00602 PMid:35849791
- Maiti
A, DiNardo CD, Daver NG, Rausch CR, Ravandi F, Kadia TM. Triplet
therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated
acute myeloid leukemia. Blood Cancer J 2021; 11:25. https://doi.org/10.1038/s41408-021-00410-w PMid:33563904 PMCid:PMC7873265
- Yilmaz
M, Kantarjian H, Short NJ, Reville P, Konopleva M, Kadia T, DiNardo CD,
Borthakur G, Pemmarajyu N, Maiti A, et al. Hypomethylating agent and
venetoclax with FLT3 inhibitor "triplet" therapy in older/unfit
patients with FLT3 mutated AML. Blood Cancer J 2022; 12: 77. https://doi.org/10.1038/s41408-022-00670-0 PMid:35501304 PMCid:PMC9061716
- Yilmaz
M, Kantarjian HM, Muftuoglu M, Kadia TM, Konopleva M, Borthakur G,
DiNardo CD, Pemmaraju N, Short NJ, Alvarado Y, et al. Quizartinib with
decitabine and venetoclax (triplet) is highly active in patients with
FLT3-ITD mutated acute myeloid leukemia (AML). J Clin Oncol 2021;
39(suppl. 15): e 19019. https://doi.org/10.1200/JCO.2021.39.15_suppl.e19019
- Short
NJ, DiNardo CD, Daver N, Nguyen D, Yilmaz M, Kadia TM, Garcia-Manero G,
Issa GC, Huang X, Qiao W, et al. A triplet combination of azacitidine,
venetoclax and gilteritinib for patients with FLT3-mutated acute
myeloid leukemia: results from a phase I/II study. Blood 2021;
138(suppl. 1): 696. https://doi.org/10.1182/blood-2021-153571
- Post
SM, Ma H, Maloney P, Zhang X, Aitken M, Mak PY, Ruvoloi VR, Yasushiro
T, Kozaki R, Chan Le, et al. AXL/MERTK inhibitor ONO-7475 potently
synergizes with VEN and overcomes VEN resistance to kill FLT3-ITD acute
myeloid leukemia. Haematologica 2022; 107: 1311-1322. https://doi.org/10.3324/haematol.2021.278369 PMid:34732043 PMCid:PMC9152975
- Janssen
M, Schmidt C, Bruch PM, Blank MF, Rohde C, Waclawiezek A, Heid D,
Rendey S, Golluer S, Vierbam L, et al. Venotoclax synergizes with
gilteritinib in FLT3 wild type high-risk AML by suppression of MCL1.
Blood 2022; in press. https://doi.org/10.1182/blood.2021014241 PMid:35857899
- Ma
J, Zhao S; Qiao X, et al. Inhibition of BCL-2 synergistically enhances
the antileukemic activity of midostaurin and gilteritinib in
preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer
Res 2019; 68: 15-26. https://doi.org/10.1158/1078-0432.CCR-19-0832 PMid:31320594 PMCid:PMC6858954
- Lachowiez
CA, Loghavi S, Furudate K, Montalban-Bravo G, Maiti A, Kadia T, Dever
N, Borthakur G, Pemmaraju N, Sasaki K, et al. Impact of splicing
mutations in acute myeloid leukemia treated with hypomethylating agents
combined with venetoclax. Blood Adv 2021; 5: 2173-2183. https://doi.org/10.1182/bloodadvances.2020004173 PMid:33885753 PMCid:PMC8095152
- Nechiporuk
T, Kurtz SE, Nikolova O, Liu T, Jones CL, D'Alessandro A, Culp-Hill R,
D'Almeida A, Joshi SK, Rosenberg M, et al. The TP53 apoptotic network
is a primary mediator of resistance to BCL2 inhibition in AML cells.
Cancer Discover 2019; 9: 910-925. https://doi.org/10.1158/2159-8290.CD-19-0125 PMid:31048320 PMCid:PMC6606338
- Aldoss
I, Zhang J, Pillai R, et al. Venetoclax and hypomethylating agents in
TP53-mutated acule myeloid leukemia. Br J Haematol 2019; 187: e45-e48. https://doi.org/10.1111/bjh.16166 PMid:31441045 PMCid:PMC6786919
- Kim
K, Maiti A, Kadia TM, Ravandi F, Daver N, Pemmaraju N, Borthakur G,
Bose P, Issa GC, Short NJ, et al. Outcomes of TP53-mutant acue myeloid
leukemia with venetoclax and decitabine. Blood 2020; 136(suppl.1):
33-36. https://doi.org/10.1182/blood-2020-140182
- Pollyea
DA, Pratz KW, Wei AH, Pullarkat V, Jonas BA, Recher C, Babu S, Sehuh
AC, Dail M, Sun Y, et al Outomes in patients with poor-risk
cytogenetics with or without TP53 mutations treated with venetoclax and
azacitidine. Clin Cancer Res 2022; in press. https://doi.org/10.1158/1078-0432.CCR-22-1183 PMid:36007102
- Shapiro
LC, Mantzaris I, Shashi A, Sica AR, Bachier-Rodriguez L, Kernblum N,
Grisham K, Feldman EJ, Verma AK, Brauynschweig I, et al. Low-dose
weekly decitabine and venetoclax in TP53-mutated myeloid malignancies.
J Clin Oncol 2022; 40(supplk. 15): e19005. https://doi.org/10.1200/JCO.2022.40.16_suppl.e19005
- Gaidzik
VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J,
Wallrabenstein T, Kolbinger B, Kohne CH, Horst HA, et al. RUNX1
mutations in acute myeloid leukemia are associated with distinct
clinic-pathologic and genetic features. Leukemia 2016; 30(11):
2160-2168. https://doi.org/10.1038/leu.2016.126 PMid:27137476
- Quesada
AE, Montalban-Bravo G, Luthra R, Patel KP, Sasaki K,Bueso-Rumos CE,
Khoury JD, Routbort MJ, Bassett R, Hidago-Lopez JE, et al.
Clinico-pathologic characteristics and outcomes of the World Health
Organization /WHO) provisional entity de novo acute myeloid leukemia
with mutated RUNX1. Mod Pathol 2020; 33: 1678-1689. https://doi.org/10.1038/s41379-020-0531-2 PMid:32238878
- Cherry
EM, Abbott D, Amaya M, McMahon C, Schwartz M, Rosser J, Sato A,
Schoiwinsky J, Inguva A, Minhajuddin M, et al. Venetoclax and
azacytidine compared with induction chemotherapy for newly diagnosed
patients with acute myeloid leukemia. Blood Adv 2021; 5(24): 5565-5573.
https://doi.org/10.1182/bloodadvances.2021005538 PMid:34610123 PMCid:PMC8714726
- Venugopal
S, DiNardo CD, Loghavi S, Qiao W, Ravandi F, Konopleva M, Kadia T,
Bhalla K, Jabbour E, Issa GC, et al. Differential prognostic impact of
RUNX1 mutations according to frontline therapy in patients with acute
myeloid leukemia. Am J Hematol 2022;Sep 10. doi: 10.1002/ajh.26724 https://doi.org/10.1002/ajh.26724 PMid:36087091
- Morsia
E, McCullough K, Joshi M, Cook J, Atkhateeb HB, Al-Kali A, Begna K,
Elliott M, Hogan W, Litzow M, et al. Venetoclax and hypomethylating
agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am
J Hematol 2020; 95: 1511-1521. https://doi.org/10.1002/ajh.25978 PMid:32833294
- Cai
X, Gao L, Teng L, Ge J, Oo ZM, Kumar AR, Gilliland DG, Mason PJ, Tan K,
Speck NA. Runx1 deficiency decreases ribosome biogenesis and confers
stress resistance to hematopoietic stem and progenitor cells. Cell Stem
Cell 2015; 17: 165-177. https://doi.org/10.1016/j.stem.2015.06.002 PMid:26165925 PMCid:PMC4530029
- Mill
CP, Fiskus W, DiNardo CD; Birdwell C, Davis JA, Kadia TM, Takahashi K,
Short N, Daver N, Ohanian M, et al. Effective therapy for AML with
RUNX1 mutation by cotreatment with inhibitors of protein translation
and BCL2. Blood 2022; 139(6): 907-921. https://doi.org/10.1182/blood.2021013156 PMid:34601571
- Vergez
F, Largeaud L, Bertoli S, Nicolau ML, Riea JB, Vergnolle I, Saland E,
Sarry A, Huguet F, et al. Phenotypically-defined stages of leukemia
arrest predict main driver mutation subgroups, and outcome in acute
myeloid leukemia. Blood Cancer J 2022; 12(9): 117. https://doi.org/10.1038/s41408-022-00712-7 PMid:35973983 PMCid:PMC9381519
- Masarova
L, DiNardo CD, Bose P, Pemmaraju N, Daver NG, Kadia TM, Chifotides HT,
Zhou L, Borthakur G, Estrov Z, et al. Single-center experience with
venetoclax combinations in patients with newly diagnosed and relapsed
AML evolving from MPNs. Blood Adv 2021; 5: 2156-2161. https://doi.org/10.1182/bloodadvances.2020003934 PMid:33885751 PMCid:PMC8095138
- Pullarkat
V, Pratz K, Dohner H, Recher C, Thirman MJ, DiNardo CD, Fenaux P, Schuh
AC, Wei AH, Pigneux A, et al. Venetoclax and azacytidine combination in
chemotherapy ineligible untreated patients with therapy-related myeloid
neoplasms, antecedent myelodysplastic syndromes, or
myelodysplastic/myeloproliferative neoplasms. J Clin Oncol 2021;
(suppl. 15): 7011. https://doi.org/10.1200/JCO.2021.39.15_suppl.7011
- Short
NJ, Venugopal S, Qiao W, Kadia TM, Ravandi F, Macaron W, Dinardo CD,
Daver N, Konopleva M, Borthakur G, et al. Impact of frontline treatment
approach on outcomes in patients with secondary AML with prior
hypomethylating agent exposure. J Hematol Oncol 2022; 15:12. https://doi.org/10.1186/s13045-022-01229-z PMid:35093134 PMCid:PMC8800349
- Ganan-Gomez
I, Yang H, Ma F, Montalban-Bravo G, Thongon N, Marchica V,
Richard-Carpentier G, Chien K, Manyam G, Wang F, et al. Stem cell
architecture drives myelodysplatic syndrome progression and predicts
response to venetoclax-based therapy. Nat Med 2022; 28: 557-567. https://doi.org/10.1038/s41591-022-01696-4 PMid:35241842 PMCid:PMC8938266
- Ball
BJ, Famulare CA, Stein EM, Tallman MS, Derkach A, Roshal M, Gill SI,
Manning BM, Koprivnikar J, McClaskey J, et al. Venetoclax and
hypomethylating agents induce high response rates in MDS, including
patients after HMA failure. Blood Adv 2020; 4(13): 2866-2870. https://doi.org/10.1182/bloodadvances.2020001482 PMid:32589727 PMCid:PMC7362378
- Maiti
A, Konopleva M. How we incorporate venetoclax in treatment regimens for
acute myeloid leukemia. The Cancer Journal 2022; 28: 2-13. https://doi.org/10.1097/PPO.0000000000000567 PMid:35072368 PMCid:PMC8785772
- Lchowiez
CA, Atluri H, DiNardo CD. Advancing the standard: venetoclax combined
with intensive induction and consolidation therapy for acute myeloid
leukemia. Ther Adv Hematol 2022; 13: 1-15. https://doi.org/10.1177/20406207221093964 PMid:35510212 PMCid:PMC9058453
- Diao
S, Nichols ED, DiNardo CD, Konopleva M, Ning J, Qiao W, Maiti A,
DiPippo AJ. Incidence of tumor lysis syndrome in patients with acute
myeloid leukemia undergoing low-intensity induction with venetoclax. Am
J Hematol 2021; 96(3): E65-E68. https://doi.org/10.1002/ajh.26060 PMid:33259075
- Arora
S, Zainaldin C, Bathini S, Gupta U, Woth S, Bachiashvili R, Godby K,
Jamy O, Rangaraju S, Diamond B, et al. Tumor lysis syndrome and
infectious complications during treatment with venetoclax combined with
azacytidine or decitabine in patients with acute myeloid leukemia. Leuk
Res 2011; 117: 106844. https://doi.org/10.1016/j.leukres.2022.106844 PMid:35487134
- Taurino
D, Frigeni M, Grassi A, Cavallaro G, Salmoiraghi S, Spinelli O,
Rambaldi A, Lussana F. Concurrent diagnosis of acute myeloid leukemia
and symptomatic COVID-19 infection: a case report successfully treated
with azacytidine-venetoclax combination.Med J Hemat Infect Dis 2021;
13(1): e2021057. https://doi.org/10.4084/MJHID.2021.057 PMid:34527209 PMCid:PMC8425345
- Cristiano
A, Palmieri R, Fabiani E, Ottone T, Divona M, Salvi A, Buccisano F,
Maurillo L, Tarella C, Arcese W, Voso MT. The venetoclax/azacitidine
combination targets the disease clone in acute myeloid leukemia, being
effective and safe in a patient with COVID-19. Med J Hemat Infect Dis
2022; 14(1): 2022041. https://doi.org/10.4084/MJHID.2022.041 PMid:35615323 PMCid:PMC9083951
- Agarwal
SK, Hu B, Chien D, Wong SL, Salem AH. Evaluation of rifampin's
transporter inhibitory and CYP3A4 inductive effects on the
pharmacokinetics of venetoclax, a BCL-2 inhibitor: results of a single-
and multiple-dose study. J Clin Pharmacol 2016; 56: 1335-1343 https://doi.org/10.1002/jcph.730 PMid:26953185
- Agarwal
SK, Salem AH, Danilov AV, Hu B, Puvvada S, Gutierrez M, Chien D, Lewis
LD, Wong SL. Effect of ketoconazole, a strong CYP3A4 inhibitor, on the
pharmacokinetics of venetoclax, a BCL-2inhibitor, in patients with
non-Hodgkin lymphoma. Br J Pharmacol 2016; 83: 846-854. https://doi.org/10.1111/bcp.13175 PMid:27859472 PMCid:PMC5346863
- Liu
H, Michmerhuizen MJ, Lao Y, Wan K, Salem AH, Sawicki J, Serby M,
Vaidyanathan S, Wong SL, Agarwal S, et al. Metabolism and disposition
of a novel B-cell lymphoma-2 inhibitor venetoclax in humans and
characterization of its unusual metabolites. Drug Met Disposition 2017;
45: 294-305. https://doi.org/10.1124/dmd.116.071613 PMid:27993930
- Roberts
AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gericitano JF, Kipps TJ,
Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with
venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016;
374: 311-322. https://doi.org/10.1056/NEJMoa1513257 PMid:26639348 PMCid:PMC7107002
- Salem
AH, Agarwal SK, Dunbar M, Enschede SHL, Humerickhouse RA, Wong SL.
Pharmacokinetics of venetoclax, a novel BCL-2 inhibitor, in patients
with relapsed or refractory chronic lymphocytic leukemia or
non-Hodgkin's lymphoma. J Clin Pharmacol 2017; 57: 484-492. https://doi.org/10.1002/jcph.821 PMid:27558232
- Agarwal
SK, DiNardo CD, Potluri J, Dunbar M, Kantarjian HM, Humerickhouse RA,
Wong SL, Menon RM, Konopleva M and Salem AH. Management of
venetoclax-posaconazole interaction in acute myeloid patients:
evaluation of dose adjustments. Clin Ther 2017; 39: 359-367. https://doi.org/10.1016/j.clinthera.2017.01.003 PMid:28161120
- Bhatnagar
S, Mukherjee D, Salem AH, Miles D, Menon RM, Gibbs JP. Dose adjustment
of venetoclax when co-administered with Posaconazole: clinical
drug-drug interaction predictions using a PBPK approach. Cancer
Chemother Pharmacol 2021; 87: 465-474. https://doi.org/10.1007/s00280-020-04179-w PMid:33398386
- Rausch
CR, DiNardo C, Maiti A, Jammal N, Kadia TM, Marx K, Borthakur GK, Savoy
M, Pemmaraju N, Marx K, et al. Venetoclax dosing in combination with
antifungal agents: real world experience in patients with acute myeloid
leukemia. Blood 2019; 134 (suppl.1): 2640. https://doi.org/10.1182/blood-2019-131988
- Rausch
CR, DiNardo C, Maiti A, Jammal N, Kadia TM, Marx K, Borthakur GK, Savoy
M, Pemmaraju N, Marx K, et al. Duration of cytopenias with concomitant
venetoclax and azole antifungals in acute myeloid leukemia. Cancer
2021; 127: 2489-2499 https://doi.org/10.1002/cncr.33508 PMid:33793970 PMCid:PMC8249340
- Eisenmann
ED, Garrison DA, Talebi Z, Jin Y, Silvaroli JA, Kim JG, Sparreboom A,
Savona MR, Mims AS and Baker SD. Interaction of antifungal drugs with
CYP3A- and OATP1B-mediated venetoclax elimination. Pharmaceutics 2022;
14: 694. https://doi.org/10.3390/pharmaceutics14040694 PMid:35456528 PMCid:PMC9025810
- Ong
F, Kim K, Konopleva MY. Venetoclax resistance: mechanistic insights and
future strategies. Cancer Drug Resistance 2022; 5: 380-400. https://doi.org/10.20517/cdr.2021.125 PMid:35800373 PMCid:PMC9255248
- Bhatt
S, Pioso MS, Olesinski EA, Yilma B, Ryan JA, Mashaka T, Leutz B, Adamia
S, Zhu H, Kuang Y, et al. Reduced mitochondrial apoptotic priming
drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer
Cell 2020; 38: 872-890. https://doi.org/10.1016/j.ccell.2020.10.010 PMid:33217342 PMCid:PMC7988687
- Pan
R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, Andreef M,
Ruvolo PP. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor
(-)BI97D6 overcomes ABT-737 resistance in acute myeloid cells. Blood
2015; 126(3): 363-372. https://doi.org/10.1182/blood-2014-10-604975 PMid:26045609 PMCid:PMC4504949
- Lin
KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley
JP, Wood KC. Targeting MCL-1/BCL-XL forestalls the acquisition of
resistance to ABT-199 in acute myeloid leukemia. Sci Rep 2016; 6:
27696. https://doi.org/10.1038/srep27696 PMid:27283158 PMCid:PMC4901329
- Ewald
L, Dittmann J, Vogler M, Fulda S. Side-by-side comparison of
BH3-mimetics identifies MCL-1 as a key therapeutic target in AML. Cell
Death Dis 2019; 10(12): 917. https://doi.org/10.1038/s41419-019-2156-2 PMid:31801941 PMCid:PMC6892884
- Hormi
M, Birsen R, Belhadj M, Huynh T, Aquilar LC, Grignano E, Hallaoui L,
Guilloneau F, Mayeux P, Hunault M, et al. Pairing MCL-1 inhibition with
venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur
J Haematol 2020; 105(5): 588-596. https://doi.org/10.1111/ejh.13492 PMid:32659848
- Ramsey
HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, Boyd KL,
Strickland SA, Sensitaffar J, Hoghdal LJ, et al. A novel MCL1 inhibitor
combined with venetoclax rescues venetoclax-resistant acute myelogenous
leukemia. Cancer Discover 2018; 8(12): 156-1581. https://doi.org/10.1158/2159-8290.CD-18-0140 PMid:30185627 PMCid:PMC6279595
- Moujalled
D, Pomilio G, Chiurau C, Ivey A, Salmon J, Rijal S, Macraild S, Zhang
L, The TC, Tong JS, et al. Combining BH3-mimetics to target both BCL-2
and MCL-1 has potent activity in pre-clinical models of acute myeloid
leukemia. Leukemia 2019; 33(4): 905-917. https://doi.org/10.1038/s41375-018-0261-3 PMid:30214012 PMCid:PMC6484700
- Carter
BZ, Mak PY, Tag W, Zhang Q, Ruvolo V, Kuruvilla VM, Wang X, Max DH,
Battula VL, Konopleva M, eal. Maximal activation of apoptosis signaling
by cotargeting antiapoptotic proteins in BH3 mimetic-resistant AML and
AML stem cells. Mol Cancer Ther 2022; 21(6): 879-889. https://doi.org/10.1158/1535-7163.MCT-21-0690 PMid:35364607
- Liu
F, Zhao Q, Su Y, Lv J, Gai Y, Liu S, Liu H, Wang Y, Wang G. Cotargeting
of Bcl-2 and Mcl-1 shows promising antileukemic activity against AML
cells including those with acquired cytarabine resistance. Exp Hematol
2022; in press. https://doi.org/10.1016/j.exphem.2021.10.006 PMid:34767916
- Weller
S, Toennieben A, Schaefer B, Beigl T, Muenchow A, Bopple K, Hofmann U,
Gillissen B, Aulitzky WE, Kopp HG, et al. The BCL-2 inhibitor
ABT-199/venetoclax synergizes with proteasome inhibition via
transactivation of the MCL-1 antagonis NOXA. Cell Daeth Discov 2022;
8(1): 215. https://doi.org/10.1038/s41420-022-01009-1 PMid:35443750 PMCid:PMC9021261
- Zhang
H, Nakauchi Y, Kohnke T, Stafford M, Bottomly D, Thomas R, Wilmot B,
McWeeney SK, Majeti R, Tyner JM. Integrated analysis of patients
samples identifies biomarkers for venetoclax efficacy and combination
strategies in acute myeloid leukemia. Nat Cancer 2020; 1(8): 826-839. https://doi.org/10.1038/s43018-020-0103-x PMid:33123685 PMCid:PMC7591155
- Pan
R, Ruvolo V, Um H, Leverson JD, Nichols G, Reed JC, Konopleva M,
Andreef M. Synthetic lethality of combined Bcl-2 inhibition and p53
activation in AML: mechanisms and superior antileukemic efficacy.
Cancer Cell 2017; 32: 748-760. https://doi.org/10.1016/j.ccell.2017.11.003 PMid:29232553 PMCid:PMC5730338
- Zhang
Q, Riley-Gillis B, Han L, Jia Y, Lodi A, Zhang H, Ganesan S, Pan R,
Konoplev SN, Sweeney SR, et al. Activation of RAS/MAPK pathway confers
MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in
acute myeloid leukemia. Signal Transd Targeted Therapy 2022; 7: 51. https://doi.org/10.1038/s41392-021-00870-3 PMid:35185150 PMCid:PMC8858957
- Stevens
BM, Joenes CI, Winters A, Dugan J, Abbott D, Savona MR, Fesik SW,
Pollyea DA, Jordan CT. PTPN11 mutations confer unique metabolic
properties and increase resistance to venetoclax and azacytidine in
acue myelogenous leukemia. Blood 2018; 132: 909. https://doi.org/10.1182/blood-2018-99-119806
- Gemans
BF, Zwaan CM, Martinelli S, Harrell P, de Lange D, Carca C, Reinhardt
D, Hahlen K, Creutzig U, Tartaglia M, et al. Differences in the
prevalence of pTPN11 mutations in FABM5 pediatric acute myeloid
leukemia. Br J Haematol 2005; 130: 800-801. https://doi.org/10.1111/j.1365-2141.2005.05685.x PMid:16115145
- Pei
S, Pollyea DA; GustafsonA, Stevens BM, Minhajuddin M, Fu R, Riemondy
KA, Gillen AE, Sheridan RM, Kim J, et al. Monocytic subclones confer
resistance to venetoclax-based therapy in patients with acute myeloid
leukemia. Cancer Discov 2020; 10: 536-551 https://doi.org/10.1158/2159-8290.CD-19-0710 PMid:31974170 PMCid:PMC7124979
- Han
L, Zhang Q, Dail M, Shi C, Cavazos A, Ruvolo VR, Zhao Y, Kim E, Rahmani
M, Mak DC, et al. Concomitant targeting of BCL2 with venetoclax and
MAPK signaling with cobimetinib in acute myeloid leukemia models.
Haematologica 2020; 105(3): 697-707. https://doi.org/10.3324/haematol.2018.205534 PMid:31123034 PMCid:PMC7049339
- Pullarkat
VA, Lacayo NI, Jabbour E, Rubnitz JE, Bajel A, Laetsch TW, Leonard J,
Colace SI, Khaw SL, Fleming KA, et al. Ventoclax and navitoclax in
combination with chemotherapy in patients with relapsed or refractory
acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer
Discovery 2021; 1(6): 1440-1453. https://doi.org/10.1158/2159-8290.CD-20-1465 PMid:33593877 PMCid:PMC9533326
[TOP]