Maria Laura Bisegna1, Maria Assunta Limongiello2, Stefano Fiori3, Irene Della Starza1, Maria Stefania De Propris1 and Stefania Trasarti1.
1 Hematology, Department of Translational and Precision Medicine, Sapienza University, Rome, Italy.
2
Dipartimento di Diagnostica per Immagini, Radioterapia Oncologica ed
Ematologia, Fondazione Policlinico Universitario A. Gemelli IRCCS,
Roma, Italy.
3 Division of Hematopathology, IEO, European Institute of Oncology IRCCS, 20141 Milan, Italy.
Correspondence to: Dr.
Maria Laura Bisegna, MD, Hematology, Department of Translational and
Precision Medicine, Sapienza University of Rome, Via Benevento 6, 00161
Rome, Italy. E-mail:
marialaura.bisegna@uniroma1.it
Published: January 1, 2023
Received: September 28, 2022
Accepted: December 22, 2022
Mediterr J Hematol Infect Dis 2023, 15(1): e2023010 DOI
10.4084/MJHID.2023.010
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
To the editor
Heavy
chain diseases (HCDs) are rare B-cell neoplasms characterized by the
production of a monoclonal immunoglobulin composed of the only heavy
chain without corresponding light chains.
These alterations are
caused by the loss of a large portion of the constant region C1 domain
of the heavy chain, which prevents the normal link to the light chain.[1]
Monoclonal heavy chains do not result in a monoclonal peak at routine
serum protein electrophoresis and are detectable only by
immunofixation. The 2017 World Health Organization (WHO) classification
of tumors of hematopoietic and lymphoid tissues includes three variants
of HCD: gamma-HCD (with IgG production), mu-HCD (with IgM production),
and alfa-HCD (with IgA production). Gamma-HCD (γHCD), also known as
Franklin's disease, is a mature B-cell neoplasm, usually with
plasmacytic differentiation, first described by Franklin in 1964. It is
a rare adult disease that may involve the most common lymph nodes, bone
marrow, and spleen. Histologically γHCD has a broad morphologic
spectrum and may resemble various lymphoproliferative disorders. Some
cases are difficult to classify and do not fall into a distinct
WHO-defined entity. Most patients show systemic symptoms such as
anorexia, weakness, fever, weight loss, recurrent bacterial infections,
organomegaly, and increasing circulating lymphocytes and plasma cells.
Autoimmune manifestations are also common (25-70% of cases), frequently
preceding the diagnosis of gamma-HCD, and they are maybe due to an
autoimmune response stimulating the capability of the abnormal gamma
heavy chain. The clinical course is highly variable, from indolent to
rapidly progressive, depending on the extension of the disease
(localized or disseminated). For this reason, treatment was reserved
for symptomatic patients in the largest series of HCD.[2] We present the
case of a patient with γHCD and concomitant T- cell large granular
lymphocyte (T-LGL) disorder. Other six similar cases were described in
the literature before.[3-5] A 65-year-old woman came to our observation
because of pancytopenia at routine blood tests, with hemoglobin 9.6
g/dL, white blood cells 4.78 x109/L, neutrophils 1.6 x109/L,
lymphocytes 2.47x109/L and platelets 47 x109/L. She complained of mild
widespread joint pain, especially in the upper limbs and pelvis. LDH
levels and liver and kidney function were normal and physical
examination was negative for superficial adenopathy or splenomegaly. A
blood smear showed an increase in LGL, equal to 42% of total
lymphocytes, while PB flow cytometry revealed 9% B-lymphocytes CD19+,
CD20+, CD22+, CD200+, CD38+, and CD5- with a small aberrant population
lacking expression of light chains (5% of total lymphocytes). Natural
killer (NK) and T-NK cells were 1% and 3%, respectively. T4/T8 ratio
was about 1.64%. Bone marrow (BM) aspirate showed an increase in
lymphocytes (34%) and plasma cells (8%), along with preserved
hemopoietic maturation and absence of blasts. The cytogenetic study
displayed a normal female karyotype. BM biopsy identified 20%
interstitial infiltrate of small B-lymphocytes
CD20+/CD5-/CD23-/CyclinD1-/DBA.44-/IgM-/IgD along with mature plasma
cells CD138 and IgG positive and negative for IgM, IgD and both kappa
and lambda light chains, similarly as observed with flow cytometry
(Figure 1a). Moreover, an additional 10% interstitial and
intrasinusoidal T-cell infiltrate CD3+, CD8+, CD57+/-, and CD4-
consistent with T-LGL expansion was found (Figure 1b). Molecular
analysis on PB and BM showed a monoclonal rearrangement of the T-cell
receptor gamma chain gene (TCR), confirming the clonal nature of the
T-LGL component and, accordingly to PB morphologic and immunophenotypic
data, the diagnosis of T-LGL lymphoproliferative disorder. Serum
electrophoresis showed hypo-gammaglobulinemia at 0.35 g/dL with
increased alfa-2-region at 1.6 g/dL but no clear monoclonal component
(Figure 3a). IgA and IgM levels were reduced, while about IgG, the
subclasses IgG1, IgG2, and IgG3 were increased, with a normal level of
IgG4. Nevertheless, serum immunofixation identified a monoclonal IgG
heavy chain without corresponding light chains (Figure 3b), also
present in the urine, according to flow cytometric analysis. A total
body CT scan resulted negative for lymphadenopathy and
hepatosplenomegaly. Due to the clinical history of arthralgia, the
patient was tested for autoantibodies: only a low titer of IgM
cryoglobulin was found, while ANA, ENA, C3, C4, anti-ccp, and anti-DNA
native resulted negative. The final diagnosis was gamma-HCD with
concomitant T-LGL disorder. For the lack of symptoms, the patient was
kept under watchful control without any specific treatment. In February
2022, our patient developed a COVID-19 infection but remained
asymptomatic despite the immunosuppression linked to his disease. At 36
months after diagnosis, she only suffers mild widespread joint pain. We
observed spontaneous improvement of PB cells count (hemoglobin 10.3
g/dL, white blood cells 5.46 x109/L, neutrophils 2 x109/L, lymphocytes
2.91 x109/L, platelets 198 x109/L), normal renal function, increased
24-hour proteinuria and quite stable IgG values at 3 g/dL. The
occurrence of γHCD and, even more, the association with T-LGL is rare.
Our literature review identified only six patients with γHCD and
concomitant T-LGL disorder, one of which was without clinical and
biological description.[3-5] T-LGL is a persistent (more than six months)
clonal expansion of cytotoxic T-cells that can arise as a primitive
disease or associate with autoimmune disorders and/or B cell
lymphoproliferative diseases.[6-9] As shown in Table 1, they were
middle-aged adults investigated to study cytopenia, splenomegaly, or
lymphadenopathy. Clinically, patients have either claimed few or
several systemic symptoms, and histologically, there was a broad
morphologic spectrum. The therapeutic approach used in the cases
reported depends on clinical and histological findings with a favorable
outcome. Cytopenia, more precisely neutropenia, is the most common
clinical finding, but up to 60% of cases are asymptomatic. Moderate
splenomegaly is frequent, while lymphadenopathy is very rare. The
clinical course is usually indolent and non-progressive; since the
association of γHCD and T-LGL disorder is exceptional, currently, there
are no defined therapeutic strategies; “watch and wait” could be a
valid attitude with accurate clinical observation, especially for
asymptomatic patients. Currently, the prognosis of these patients
remains unclear. However, the somatic mutations on genes involved in
the pathogenesis of these entities (i.e., STAT3) may play a role in the
course of the disease.[10] In conclusion, we report a rare association of
two lymphoproliferative diseases, one with B phenotype and the other
with T phenotype. As postulated in previous reports, it may be a
non-casual coexistence since it could result from strong common antigen
stimulation mediated by an infectious agent or autoantigen. However,
the exact etiopathogenetic substratum is still to be clarified. Further
investigations of the pathogenesis of γHCD with or without T-LGL
disorders in a larger study cohort could be of value.
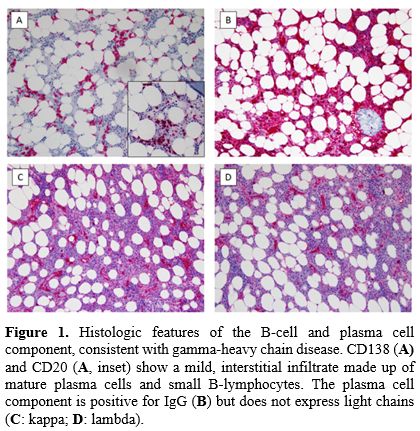 |
Figure 1. Histologic features of the B-cell and plasma cell component, consistent with gamma-heavy chain disease. CD138 (A)
and CD20 (A, inset) show a mild, interstitial infiltrate made up of
mature plasma cells and small B-lymphocytes. The plasma cell component
is positive for IgG (B) but does not express light chains (C: kappa; D: lambda). |
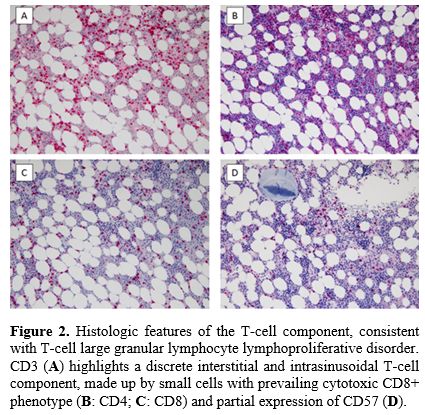 |
Figure 2. Histologic
features of the T-cell component, consistent with T-cell large granular
lymphocyte lymphoproliferative disorder. CD3 (A)
highlights a discrete interstitial and intrasinusoidal T-cell
component, made up by small cells with prevailing cytotoxic CD8+
phenotype (B: CD4; C: CD8) and partial expression of CD57 (D).
|
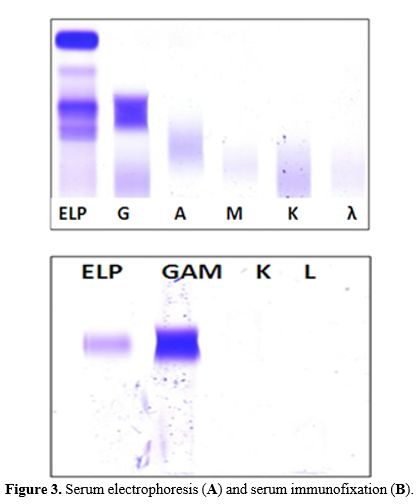 |
Figure 3. Serum electrophoresis (A) and serum immunofixation (B).
|
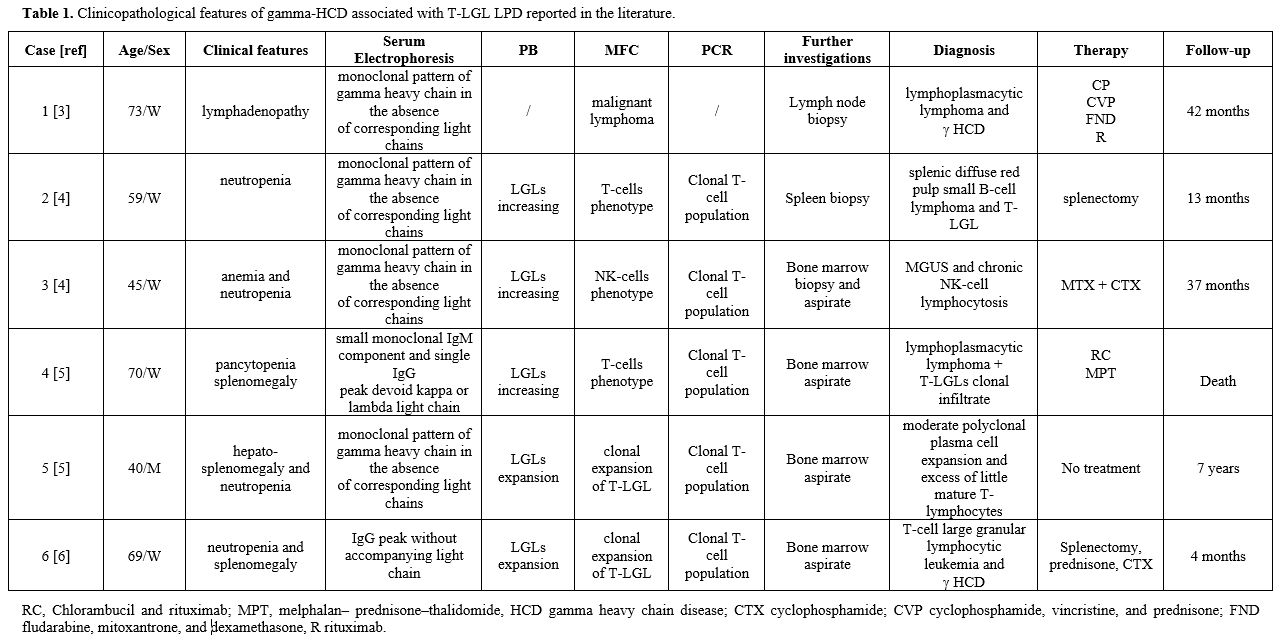 |
Table 1. Clinicopathological features of gamma-HCD associated with T-LGL LPD reported in the literature.
|
Author contributions
MLB
and MAL wrote the manuscript; SF performed cytological and
immunohistochemical analysis and collected samples and figures; IDS
collected molecular data; MSDP performed flow-cytometric,
electrophoresis, and immunofixation analysis; ST designed the research
study, collected patient data, and revised the manuscript.
References
- Bianchi G, Anderson KC, Harris NL and Sohani AR. The
heavy chain diseases: clinical and pathologic features. Oncology. 2014;
28:45-53.
- Wahner-Roedler DL, Witzig TE, Loehrer LL and Kyle
RA. Gamma-heavy chain disease: review of 23 cases. Medicine.
2003;82(4):236-50. https://doi.org/10.1097/01.md.0000085058.63483.7f PMid:12861101
- Bieliauskas
S, Tubbs RR, Bacon CM, Eshoa C, Foucar K, Gibson SE, Kroft SH, Sohani
AR, Swerdlow SH and Cook JR. Gamma heavy-chain disease: defining the
spectrum of associated lymphoproliferative disorders through analysis
of 13 cases. Am J of Sur Pathol. 2012; 36(4):534-43.
https://doi.org/10.1097/PAS.0b013e318240590a PMid:22301495
PMCid:PMC3715127
- Wahbi A, Neel A, Perrin F, Graveleau J, Mahe B,
Dejoie T and Hamidou M. Gamma heavy chain disease associated with large
granular lymphocytic leukemia: A report of two cases and review of the
literature. Hematology. 2016;21(2):92-4.
https://doi.org/10.1179/1607845415y.0000000037 PMid:26222587
- Zhang
L, Sotomayor EM, Papenhausen PR, Shao H, Moscinski LC, Sandin RL,
Caceres G, Valenica H, Malafa M, List AF and Sokol L. Unusual
concurrence of T-cell large granular lymphocytic leukemia with Franklin
disease (gamma heavy chain disease) manifested with massive
splenomegaly. Leuk Lymphoma. 2013;54(1):205-8.
https://doi.org/10.3109/10428194.2012.697561 PMid:22694793
- Moss
PA, Gillespie G. Clonal populations of T-cells in patients with B-cell
malignancies Leuk Lymphoma 1997 27: 231-238. https://doi.org/10.3109/10428199709059679 PMid:9402322
- Wlodarski
MW, O'Keefe C, Howe EC, Risitano AM, Rodriguez A, Warshawsky I,
Loughran TP Jr and Maciejewski JP. Pathologic clonal cytotoxic T-cell
responses: nonrandom nature of the T-cell-receptor restriction in large
granular lymphocyte leukemia. Blood. 2005;15;106(8):2769-80.
https://doi.org/10.1182/blood-2004-10-4045 PMid:15914562
- Goyal
T, Thakral B, Wang SA, Bueso-Ramos CE, Shi M, Jevremovic D, Morice WG,
Zhang QY, George TI, Foucar KK, Bhattacharyya S, Bagg A, Rogers HJ,
Bodo J, Durkin L and Hsi ED. T-Cell Large Granular Lymphocytic Leukemia
and Coexisting B-Cell Lymphomas: A Study From the Bone Marrow Pathology
Group. Am J Surg Pathol. 2018;29;149(2):164-171.
https://doi.org/10.1093/ajcp/aqx146 PMid:29365010
- Papadaki T, Stamatopoulos K, Kosmas C, Paterakis
G, Kapsimali V, Kokkini G, Economopoulos T, Stefanoudaki-Sofianatou K,
Marinakis T, Gardikas E, Kalmantis T. Clonal T-large granular
lymphocyte proliferations associated with clonal B cell
lymphoproliferative disorders: report of eight cases. Leukemia 16,
2167-2169 (2002). https://doi.org/10.1038/sj.leu.2402643 PMid:12357377
- Rivero A,
Mozas P, Jiménez L, López-Guerra M, Colomer D, Bataller A, Correa J,
Rivas-Delgado A, Bastidas G, Baumann T, Martínez-Trillos A, Delgado J,
Giné E, Campo E, López-Guillermo A, Villamor N, Magnano L, Matutes E.
Clinicobiological Characteristics and Outcomes of Patients with T-Cell
Large Granular Lymphocytic Leukemia and Chronic Lymphoproliferative
Disorder of Natural Killer Cells from a Single Institution. Cancers
(Basel). 2021 Aug 2;13(15):3900. http://10.3390/cancers13153900 PMid:34359799 PMCid:PMC8345581