Buket Altinok Gunes1,2*, Yalda Hekmatshoar2,3*, Tulin Ozkan2, Sureyya Bozkurt4, O. Sena Erdogan Aydos2, Yahya Buyukasik4, Elifcan Aladag5 and Asuman Sunguroglu2.
1 Vocational School of Health Services, Ankara University, Ankara, Turkey.
2 Department of Medical Biology, Faculty of Medicine, Ankara University, Ankara, Turkey.
3 Department of Medical Biology, Faculty of Medicine, Altinbas University, Istanbul, Turkey.
4 Department of Medical Biology, Faculty of Medicine, Istinye University, Istanbul, Turkey.
5 Department of Hematology, Faculty of Medicine, Hacettepe University, Ankara, Turkey.
* First two author equally contributed.
Correspondence to:
Buket Altinok Gunes, PhD. Ankara University, Vocational School of
Health Services, 06290 Ankara, Turkey. Tel: 00903123573242/6642.
E-mail:
baltinok@ankara.edu.tr Orcid: 0000-0002-8852-6626
Yalda Hekmatshoar. Altinbas University, Faculty of Medicine, Department of Medical Biology, Istanbul, Turkey. E-mail:
yalda.hekmatshoar@altinbas.edu.tr Orcid: 0000-0003-4683-074X
Published: January 1, 2023
Received: October 07, 2022
Accepted: December 21, 2022
Mediterr J Hematol Infect Dis 2023, 15(1): e2023008 DOI
10.4084/MJHID.2023.008
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Chronic
myeloid leukemia (CML) is a malignant hematopoietic stem cell disease
resulting in the fusion of BCR and ABL genes and characterized by the
presence of the reciprocal translocation t(9;22)(q34;q11). BCR-ABL, a
product of the BCR-ABL fusion gene, is a structurally active tyrosine
kinase and plays an important role in CML disease pathogenesis.
Imatinib mesylate (IMA) is a strong and selective BCR-ABL tyrosine
kinase inhibitor. Although IMA therapy is an effective treatment,
patients may develop resistance to IMA therapy over time. This study
investigated the possible genetic resistance mechanisms in patients
developing resistance to IMA. We did DNA sequencing in order to detect
BCR-ABL mutations, which are responsible for IMA resistance. Moreover,
we analyzed the mRNA expression levels of genes responsible for
apoptosis, such as BCL-2, P53, and other genes (SCD-1, PTEN). In a
group of CML patients resistant to IMA, when compared with
IMA-sensitive CML patients, a decrease in SCD-1 gene expression levels
and an increase in BCL-2 gene expression levels was observed. In this
case, the SCD-1 gene was thought to act as a tumor suppressor. The
present study aimed to investigate the mechanisms involved in IMA
resistance in CML patients and determine new targets that can be
beneficial in choosing the effective treatment. Finally, the study
suggests that the SCD-1 and BCL-2 genes may be mechanisms responsible
for resistance.
|
Introduction
Chronic
myeloid leukemia (CML) is a neoplastic disease characterized by the
Philadelphia chromosome. Philadelphia chromosome results from the
reciprocal translocation between chromosomes t (9;22) (q34; q11) that
fuses the BCR and ABL genes and is present in 95% of patients with CML.[1,2] BCR-ABL fusion has a consistent tyrosine kinase activity that leads to unregulated proliferation and apoptosis inhibition.[3]
CML can be classified into three phases, chronic phase (CP),
accelerated phase (AP), and blast phase (BP). Most patients have CML in
the chronic phase when diagnosed, but if they are not given treatment,
it can advance and, after 3-5 years, reach the more deadly accelerated
phase or terminal blast crisis.
Patients with chronic phase CML typically present with an increase in
immature and mature myeloid components and retention of hematological
differentiation. Anemia, leukocytosis, splenomegaly, and infrequently
leukostasis symptoms are seen in the CP of the disease. Compared to CP,
accelerated and blast crisis CML phenotypes are more variable and
aggressive. A significant decline in cellular differentiation and the
replacement of mature cells by immature blasts defines these two
phases. More than 50% of patients have a myeloid blast stage similar to
acute myeloid leukemia (AML) during a blast crisis.[4,5]
IMA is a tyrosine kinase inhibitor (TKI) introduced as a first-line therapy for patients in all phases of CML.[6,7]
Although IMA is an efficient therapy and increases the survival rate in
patients with CML, IMA resistance becomes an obstacle in therapy,
leading to therapeutic failure and relapse.[1]
Different mechanisms are involved in IMA resistance in CML. IMA
resistance mechanisms are classified into two main groups: i) BCR-ABL
dependent and ii) BCR-ABL independent mechanisms.[8] The development of point mutations in the BCR-ABL tyrosine kinase domain[9] and overexpression of the BCR-ABL gene are the most common BCR-ABL-dependent mechanisms.[10]
BCR-ABL independent mechanisms include apoptosis inhibition,
overexpression of efflux drug transporters, DNA repair, autophagy, and
metabolic pathways.[11] Recent studies have shown
that fatty acid metabolism affects cancer development and drug
resistance. Fatty acids (FAs) are crucial for cancer cell membrane
biosynthesis and provide energy source during proliferation and
metabolic stress conditions.[12] Therefore, viable cells should sustain the ratio of saturated to unsaturated FAs to maintain membrane fluidity.
Stearoyl-CoA desaturases (SCDs) are endoplasmic reticulum enzymes
involved in FA desaturation in an oxygen-dependent manner. SCD-1 is an
important enzyme responsible for FA desaturation and is detected in all
human tissues. SCD-1 converts saturated FAs into monounsaturated FAs to
enhance unsaturated FAs levels.[13] SCD-1 is
upregulated in different cancer types, including lung, breast,
prostate, liver, and kidney, and correlates with aggressiveness and
oncogenesis.[14] According to the cancer type, SCD-1 plays different roles in regulating cell proliferation, survival, and apoptosis.[13]
In mouse models, a decreased expression level of SCD-1 regulated
apoptosis through modulation of PTEN, P53, and BCL-2 expression at the
transcriptional and/or posttranscriptional level.[13] Several studies indicate a link between P53 and PTEN as tumor suppressors and regulate cell proliferation and death.[15]
This study investigated the mechanisms that affect IMA resistance in
patients with CML and identified new targets for an effective treatment
method. We found that the expression levels of the SCD-1 gene decreased
in IMA resistance patients with CML and K562r (IMA-resistant) cell
lines. However, we discovered higher expression levels of the BCL-2
gene in patients and K562r cells compared to the control groups. The
downregulation of SCD-1 and upregulation of BCL-2 genes might be
responsible for the IMA resistance in CML independent of point
mutations in the BCR-ABL tyrosine kinase domain.
Materials and methods
Patients.
In this investigation, bone marrow/blood samples from 10 patients
responsive to IMA and 12 resistant to IMA, with CML diagnosis from the
basic oncology branch at Hacettepe University, were studied. Bone
marrow samples obtained from 5 healthy individuals were used as the
control with no previous malignant disease diagnosis. IM resistance was
documented with both cytogenetic and molecular tests. In addition, the
patients who were changed to another TKI were followed up with BCR-ABL.
Each sample was included in the study after informed consent forms were
obtained and Hacettepe University Faculty of Medicine's Institutional
Ethics Committee approved the study (approval no. GO 13/20). Bone
marrow (BM) samples were collected from the 10 patients responsive to
IMA (median age 40 years, 7 females and 3 males) and 12 patients
resistant to IMA (median age 44 years, 4 females and 8 males). The
control group comprised 5 healthy individuals (median age 35 years, 1
female and 4 males).
Gene Expression Omnibus analysis.
The SCD-1 gene expression levels were studied using the Gene expression
Omnibus dataset (GSE5550). The microarray data of GSE5550 downloaded
from the GEO database (GPL201 Affymetrix Human HG-Focus Target Array)
includes 8 healthy controls and 9 patients with newly diagnosed CML who
were untreated in the chronic phase.[16]
Cell Lines and Cell Cultures. For the IMA-resistant CML
cell line, cells from the K562 susceptible cell line obtained from
exposure to increasing IMA doses were used. K562s (IMA sensitive) and
K562r (IMA resistant) CML cell lines were incubated using RPMI 1640
medium containing 1% penicillin/streptomycin, 5% L-glutamine, and 10%
fetal bovine serum in an incubator at 37 ℃ with 5% CO2. For the K562r
cell line, a medium containing 5 µM IMA was used.
Sequence Analysis. BCR-ABL mutations responsible for
resistance to IMA (F317L, T315I, M351T, E355G, F359V, M244V, L248V,
G250E, Q252H, Y253F, E255K, M343T) were analyzed with the DNA
sequencing method. RNA isolation was performed from the samples using a
triazole reagent. From the isolated RNA samples, cDNA was synthesized
using the iScript cDNA synthesis kit (Biorad, Hercules, CA) according
to the kit protocol. Using the P210 PCR product proliferated from the
cDNA, the fourth and sixth exons were proliferated with nested PCR (Table 1). PCR samples passed through purification stages by the Promega A9281 Wizard SV Gel and PCR Clean-Up System kit
protocol (Promega, Madison, Wis., USA). After purification, cycle
sequencing was performed by the Big Dye Terminator v3.1 Cycle
sequencing kit protocol (Applied Biosystems, Foster City, CA, USA). For
DNA sequence analysis of the samples, an ABI 310 Genetic Analyzer
(Applied Biosystems) Sequence device was used. DNA sequence analysis
results were assessed in terms of mutations in each sample using the
Chromas program.
 |
- Table 1. Primer sequences used for Sequence Analysis.
|
Quantitative Real-Time PCR.
The expression levels of SCD-1, BCL-2, P53, PTEN and HPRT
(hypoxanthine-guanine phosphoribosyltransferase) genes related to IMA
resistance were analyzed quantitatively using the quantitative
real-time PCR (qRT-PCR) method. Quantitative real-time PCR was
performed using a Biorad device (Biorad, CA) at optimized temperatures
for primers (Alpha DNA, USA) using SYBR Green PCR Master Mix (iTaq
Universal SYBR Green Supermix, Biorad, CA). Experiments were performed
with 3 biological triplicates. Primer sequences are given in Table 2.
 |
- Table 2. Primer sequences used for real-time quantitative PCR.
|
Statistics.
The experimental results were all reported as mean SD (standard
deviation). GraphPad Prism statistical software was employed for the
analysis. The means of the two groups were compared by performing the
one-sample t-test, and ANOVA was used to analyze the means of numerous
samples. Statistical significance was defined as p < 0.05 and p <
0.001.
Results
Patients' clinical data.
This study included 22 patients at least 18 years old with
Philadelphia-positive chronic phase CML who followed in the Hacettepe
University Department of Hematology. The diagnosis was established
based on bone marrow examination, also cytogenetic and molecular
studies. All patients were followed for at least 3 months, and a
complete blood count and molecular study for BCR-ABL was conducted on
every visit of the patients. The chronic phase (CP) was defined
according to standard criteria.[17] Cytogenetics and
molecular responses to TKIs were rated according to the European
Leukemia Net (ELN) guidelines. The principle of the molecular response
depends upon the measurement of the BCR-ABL transcript levels relative
to a control gene.[18] All patients initiated IMA 400
mg/day as first-line treatment. IMA was maintained in 10 patients
(47.8%), while 7 (30.4%) were treated with two TKIs and the remainder
with >2 TKIs. Twelve patients who failed IMA treatment were treated
with Dasatinib or Nilotinib as second-line treatment in a median of
26.4 months (2.5-172,5). Nine patients and the remaining 3 received
Dasatinib 100 mg/day and Nilotinib 400 mg BID, respectively. A total of
5 failed Dasatinib treatments were changed to Nilotinib in a median of
19 months (13-47,4). In addition, 3 patients underwent allogeneic
hematopoietic stem cell transplantation from a human leukocyte
antigen-fully matched sibling donor.
Analysis of ABL Kinase mutations in BCR-ABL gene causing IMA resistance in patients with CML and cell lines.
The presence of mutations responsible for resistance in patients with
CML unresponsive to IMA and in K562s and K562r (5 μM) cell lines was
researched using the DNA sequence analysis method. None of the patients
resistant to IMA had mutations encountered (Figure 1). Furthermore, our previous study found no mutations in K562s and K562r cell lines.[19]
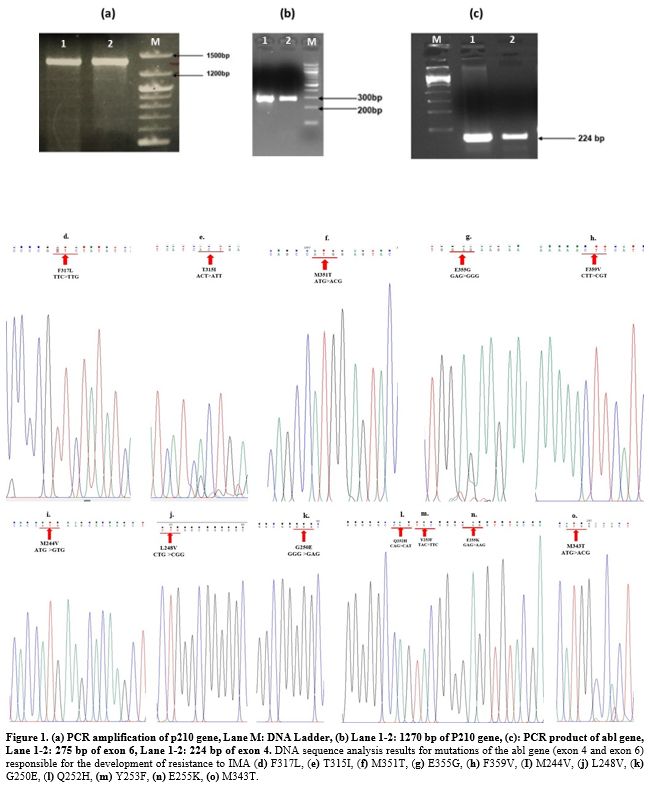 |
- Figure 1. (a)
PCR amplification of p210 gene, Lane M: DNA Ladder, (b) Lane 1-2: 1270
bp of P210 gene, (c): PCR product of abl gene, Lane 1-2: 275 bp of exon
6, Lane 1-2: 224 bp of exon 4. DNA sequence analysis results for
mutations of the abl gene (exon 4 and exon 6) responsible for the
development of resistance to IMA (d) F317L, (e) T315I, (f) M351T, (g)
E355G, (h) F359V, (I) M244V, (j) L248V, (k) G250E, (l) Q252H, (m)
Y253F, (n) E255K, (o) M343T.
|
Gene expression levels of SCD-1 in patients.
Gene expressions were determined by qRT-PCR in patients. Firstly, we
analyzed the gene expression levels of SCD-1 in all CML patients and
the control group. The SCD-1 gene expression levels increased 5-fold in
IMA-resistant patients and 8-fold in IMA-sensitive patients compared to
the control group (Figure 2).
Later, we classified CML patients into 2 groups as patients sensitive
to IM therapy and resistant to therapy, and investigated the SCD-1 gene
expression levels in these groups. As shown in Figure 2,
SCD-1 gene expressions were significantly lower in IMA-resistant CML
patients than in IMA-sensitive patients (p<0.001). Furthermore,
SCD-1 gene expression in CML patients was compared to healthy controls
using a gene expression dataset (GSE5550), and similar to the results
of our study, a notable difference was detected in SCD-1 expression
between CML patients and controls (p< 0,0003) (Figure 3).
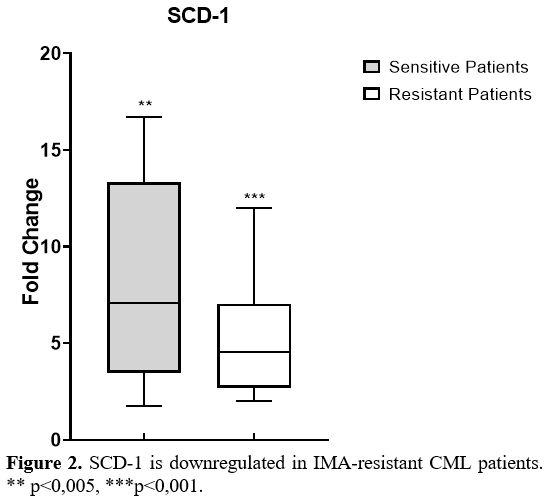 |
Figure 2. SCD-1 is downregulated in IMA-resistant CML patients. ** p<0,005, ***p<0,001. |
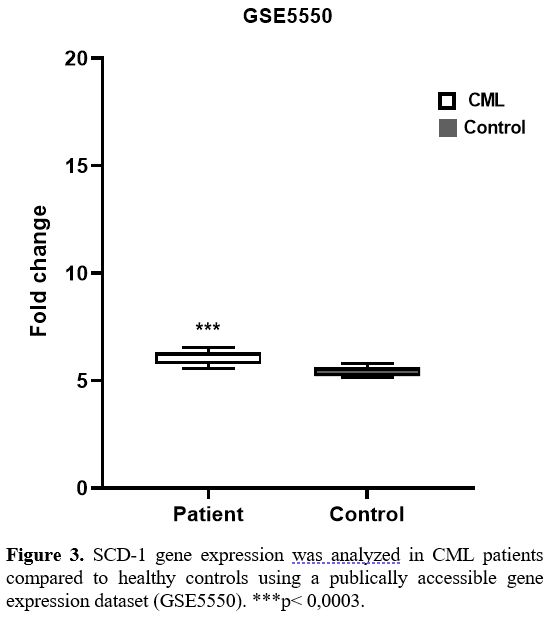 |
Figure
3. SCD-1 gene expression was analyzed in CML patients compared to
healthy controls using a publically accessible gene expression dataset
(GSE5550). ***p< 0,0003.
|
Expression levels of apoptotic genes in patients. Apoptotic gene expressions (BCL-2, PTEN, and P53) were significantly
higher in IMA-resistant CML patients than in IMA-sensitive patients (Figure 4 a, b and c).
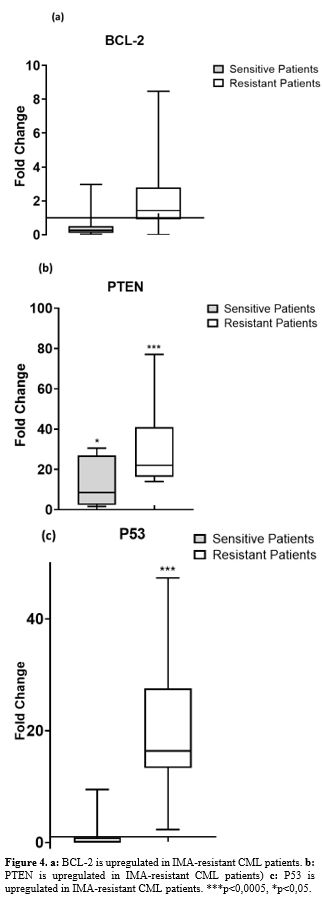 |
- Figure 4. a: BCL-2 is upregulated in IMA-resistant CML patients. b: PTEN is upregulated in IMA-resistant CML patients) c: P53 is upregulated in IMA-resistant CML patients. ***p<0,0005, *p<0,05.
|
Expressions levels of genes (SCD-1, PTEN, BCL-2) in K562s and K562r cell lines.
In K562r cells, there was a statistically significant reduction in
SCD-1 gene expression levels (p=0.0082, p<0.05) and an increase in
BCL-2 gene expression levels (p=0.0452, p<0.05) compared to K562s
cells, in parallel to the situation in IMA-resistant patients with CML (Figure 5).
No difference was identified between K562r and K562s cells for PTEN
gene expression levels. Both cell lines undergo deletion in the P53
gene region.
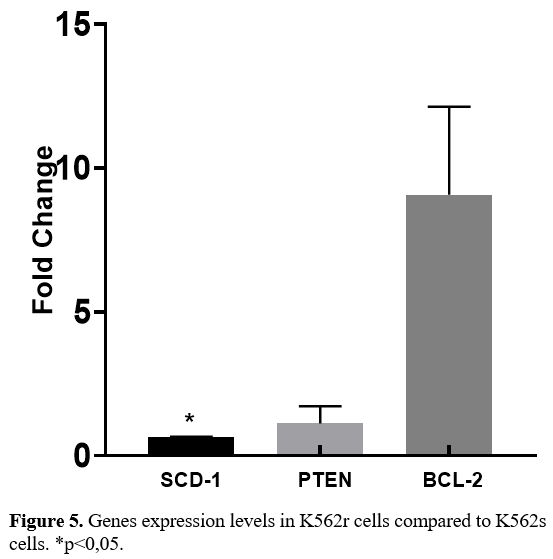 |
- Figure 5. Genes expression levels in K562r cells compared to K562s cells. *p<0,05.
|
Discussion
Chronic
myelogenous leukemia (CML) is a clonal hematopoietic disorder that
results from the balanced translocation of the ABL1 gene on chromosome
9 to the BCR region on chromosome 22, leading to the arise of the
fusion protein BCR-ABL1, which contains an activated tyrosine kinase
domain that is responsible for the clonal transformation. The
first-line standard treatment of CML is the administration of a
TKI-class drug. Imatinib is currently accepted as first–line agent with
a high impact on allowing a deep molecular response. Although imatinib
has shown very high success in CML patients, the success rate has
decreased due to resistance to imatinib. Mutations in the BCR-ABL
kinase region are the most important cause of developing resistance.
Therefore, identifying CML patients more likely to experience disease
progression or develop resistance is crucial, as this affects the
primary TKI option. The primary topics of novel findings in the field
of CML are gene expression profile, CML-leukemic stem cells,
next-generation genomics, genetic polymorphisms, multi-drug resistance
genes, and existing BCR-ABL kinase domain mutations.[20]
Our study is the first to demonstrate the role of SCD-1 in the biology
of CML and its effect on IM resistance using bone marrow samples of CML
patients. This study's results may represent a clinical significance in
CML therapy and overcoming IM resistance in patients. As so little is
known about the relationship between SCD-1, P53, and BCL-2 pathways,
there is no evidence that SCD-1 plays an active role in CML in the
literature; his involvement in CML could be an epiphenomenon.
BCR-ABL-dependent and independent molecular mechanisms have key roles
in CML drug resistance. First, we targeted BCR-ABL-dependent
mechanisms. We examined mutations using DNA sequencing for this aim,
and we found no point mutations in the BCR-ABL tyrosine kinase domain
in patients with CML resistant to IMA. These findings suggested that
BCR-ABL independent mechanisms, such as activation of alternative
signaling pathways (such as apoptotic and metabolic pathways),
influence drug resistance in CML patients.
Studies show that cancer and metabolism are tightly linked. Cancer cells display changes in fatty acid metabolism.[13]
Lipid metabolism changes impact tumor cell growth, spread, and
chemotherapeutic treatment resistance. In various cancer types, key
genes for fatty acid metabolism are defined as prognostic biomarkers
related to relapse or survival.[21] One of these key genes is SCD-1.[13]
SCD-1, a transmembrane protein in the endoplasmic reticulum (ER)
organelle, synthesizes saturated fatty acid from stearic acid and
unsaturated fatty acid oleic acid. SCD-1 is necessary to stimulate
lipid biosynthesis to ensure new phospholipids for cell membrane
biogenesis in the mitosis cell cycle process.[22,23]
The presence of SCD-1 was detected in almost every tissue, while
disrupted expression levels of this gene were identified in many cancer
types.
Several studies have proposed that SCD-1 is a new biomarker for
pulmonary cancer and that excessive expression of SCD-1 associated with
clinical symptoms is observed more frequently in patients with
advanced-stage lung cancer and causes poor prognosis.[24]
Similarly, SCD-1 levels are high in the tissues of colorectal cancer
patients, and the literature states that SCD-1 is a biomarker for
colorectal cancer and may be used for therapeutic purposes. In vivo and
in vitro pharmacological studies revealed that SCD-1 inhibition stopped
the growth of colorectal tumors through cellular ceramide production,
mitochondrial dysfunction, and induction of apoptosis in colorectal
cancer cells.[22] Recent studies showed that higher
levels of SCD-1 expression in human bladder cancers were associated
with disease stage, lymph node metastasis, and low survival percentage.[25]
High levels of SCD-1 expression predict poor prognosis in stomach
cancer patients, and the potential of Scd-1 was shown in the literature
as a biological biomarker for the early detection of stomach cancer.[26]
When clinical targeting of the SCD-1 gene with antileukemic and
anti-lymphoma treatments was studied for the first time in the
literature, the administration of Bezafibrate and Medroxyprogesterone
acetate combined therapy to acute myeloid leukemia (AML) and Burkitt
lymphoma cell lines, causing reduced SCD-1 levels, showed anticancer
activity.[27] In the literature, SCD-1 was shown to
regulate the proliferation and survival of cancer cells; at the same
time, the knock-down of SCD-1 by siRNA was stated to significantly
reduce the survival of multiple human tumor cell lines.[28]
According to a recent study, SCD-1 was shown to affect leukemic stem
cells in mice and play a critical role in CML development. Contrary to
studies conducted with various cancer types, Zhang et al. (2012) found
that SCD-1 regulated apoptosis of leukemic stem cells (LSC) and acted
as a tumor suppressor in leukemic cells. This situation shows that
SCD-1 may play different roles in different cancers. Additionally,
while the absence of SCD-1 accelerates CML development, Ph+ B-ALL
remains undisrupted. For this reason, SCD-1 has different roles in
leukemia subtypes and acts in a way specific to the cell type.
Additionally, according to Zhang et al., the deficiency of SCD-1 in
leukemic stem cells causes disrupted expression of genes associated
with apoptosis, resulting in reduced PTEN, P53, and increased BCL-2
expression. According to these results in the literature, PTEN, P53,
and BCL-2 transcriptional and/or posttranscriptional levels were shown
to be regulated by SCD-1.[13]
In our study, we first used the GEO database to identify the SCD-1 gene
expression levels of all CML patients. Second, we analyzed the mRNA
expression levels of apoptotic (BCL-2, P53) and other genes (SCD-1,
PTEN), which can be responsible for the IMA resistance of patients with
CML and K562 cells. According to the transcriptome analysis in the gene
microarray database (GSE5550), the SCD-1 gene expression levels in CML
patients were elevated compared to the control group; these data are
similar to our findings. When the IMA-resistant CML patient group with
a large LSC population was compared with the IMA-sensitive patients
with CML, there was a reduction in SCD-1 gene expression levels and an
increase in BCL-2 gene expression levels, parallel to the study by
Zhang et al. This situation suggests that the SCD-1 gene acts like a
tumor suppressor in patients with CML resistant to IMA. The
upregulation of BCL-2 gene expression in patients resistant to IMA
increases the proliferation of leukemic cells and ensures their
survival. In our study, the K562r cell line and IMA-resistant CML
patient group were found to have similar results in terms of SCD-1 and
BCL-2 gene expressions. In the literature, similar studies were
conducted to reveal the role of BCL-2 in CML. In a mouse model, when
BCL-2 was administered alone or combined with TKIs, the inhibition
effect increased, the antileukemic effect developed, and survival
durations increased. Combined inhibition of BCL-2 and BCR-ABL tyrosine
kinase was shown to have significant potential for response and
treatment rates in the chronic phase and blast crisis stages in CML.[29]
Studies with K562 cells similarly identified that BCL-2 and TKI
combined treatments prevented IMA resistance related to BCL-2 increase
or BIM or BAD loss.[30] In this situation, it was concluded that the BCL-2 gene might be a key survival factor for CML.[26]
In our study, increases were identified in PTEN and P53 expression
levels, 2 important genes playing roles in the cell cycle in patients
with CML developing resistance to IMA. Yunes et al. found that cells
carrying P53 loss of function mutations and overexpressing P53 protein
were chemotherapy-resistant and displayed increasing amounts of ABCB1
(ATP-dependent translocase) and MRP1/ABCC1 (Multidrug Resistance
Protein 1).[31] Contrary to our study, in the
literature, PTEN was shown to t as a tumor suppressor inhibiting LSCs.
Overexpression of PTEN reduced the percentage of bone marrow LSCs;
thus, it was considered that PTEN inhibited LSCs.[32,33]
The increase in PTEN gene expression levels in our patients developing
resistance to IMA leads to the consideration that the cell cycle
slowing could be studied. No significant difference was found between
the PTEN gene expression levels in the K562r and K562s cell lines.
Conclusions
In
our study, apart from ABL kinase mutations responsible for resistance,
SCD-1, and BCL-2 genes might be mechanisms responsible for resistance.
In conclusion, for the first time in this study, variations in SCD-1
gene expression were illustrated in CML patient samples. Furthermore,
increasing BCL-2 expression linked to suppressing the SCD-1 gene may
develop IMA resistance in the patient. This supports the view that IMA
and BCL-2 inhibitor combinations should be added to treatment protocols
for patients with CML. Additionally, there is a need for more research
in the future about detailed mechanisms of how SCD-1 regulates PTEN,
P53, and BCL-2 expression.
Funding
This
research was financially supported by a grant from Scientific Research
Projects of Ankara University (project no. 15B0230004).
References
- Meenakshi Sundaram, D.N., Jiang X, Brandwein JM,
Valencia-Serna J, Remant KC, Uludağ H., Current outlook on drug
resistance in chronic myeloid leukemia (CML) and potential therapeutic
options. Drug Discov Today, 2019. 24(7): p. 1355-1369. https://doi.org/10.1016/j.drudis.2019.05.007 PMid:31102734
- Asnafi,
A.A., Deris Zayeri Z, Shahrabi S, Zibara K, Vosughi T. Chronic myeloid
leukemia with complex karyotypes: Prognosis and therapeutic approaches.
J Cell Physiol, 2019. 234(5): p. 5798-5806. https://doi.org/10.1002/jcp.27505 PMid:30430567
- Cambier,
N. Chopra R, Strasser A, Metcalf D, Elefanty AG. BCR-ABL activates
pathways mediating cytokine independence and protection against
apoptosis in murine hematopoietic cells in a dose-dependent manner.
Oncogene, 1998. 16(3): p. 335-48. https://doi.org/10.1038/sj.onc.1201490 PMid:9467959
- ernandes,
A., N. Shanmuganathan, and S. Branford, Genomic Mechanisms Influencing
Outcome in Chronic Myeloid Leukemia. Cancers (Basel), 2022. 14(3). https://doi.org/10.3390/cancers14030620 PMid:35158889 PMCid:PMC8833554
- Chen, Y., et al., Molecular and cellular bases of chronic myeloid leukemia. Protein Cell, 2010. 1(2): p. 124-32. https://doi.org/10.1007/s13238-010-0016-z PMid:21203982 PMCid:PMC4875160
- Palandri,
F., Castagnetti F, Alimena G, Testoni N, Breccia M, Luatti S,
Rege-Cambrin G, Stagno F, Specchia G, Martino B, Levato L, Merante S,
Liberati AM, Pane F, Saglio G, Alberti D, Martinelli G, Baccarani M,
Rosti G.The long-term durability of cytogenetic responses in patients
with accelerated phase chronic myeloid leukemia treated with imatinib
600 mg: the GIMEMA CML Working Party experience after a 7-year
follow-up. Haematologica, 2009. 94(2): p. 205-12. https://doi.org/10.3324/haematol.13529 PMid:19144656 PMCid:PMC2635408
- Druker,
B.J, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville
R, Talpaz M., the activity of a specific inhibitor of the BCR-ABL
tyrosine kinase in the blast crisis of chronic myeloid leukemia and
acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J
Med, 2001. 344(14): p. 1038-42. https://doi.org/10.1056/NEJM200104053441402 PMid:11287973
- Nestal
de Moraes, G., Souza PS, Costas FC, Vasconcelos FC, Reis FR, Maia RC.
The Interface between BCR-ABL-Dependent and -Independent Resistance
Signaling Pathways in Chronic Myeloid Leukemia. Leuk Res Treatment,
2012. 2012: p. 671702. https://doi.org/10.1155/2012/671702 PMid:23259070 PMCid:PMC3505928
- Awidi
A, Ababneh N, Magablah A, Bsoul N, Mefleh R, Marei L, Abbasi S., ABL
kinase domain mutations in patients with chronic myeloid leukemia in
Jordan. Genet Test Mol Biomarkers, 2012. 16(11): p. 1317-21. https://doi.org/10.1089/gtmb.2012.0147 PMid:23009571 PMCid:PMC3483047
- Quintas-Cardama,
A. and J. Cortes, Molecular biology of bcr-abl1-positive chronic
myeloid leukemia. Blood, 2009. 113(8): p. 1619-30. https://doi.org/10.1182/blood-2008-03-144790 PMid:18827185 PMCid:PMC3952549
- Hekmatshoar
Y, Nakhle J, Galloni M, Vignais ML. The role of metabolism and
tunneling nanotube-mediated intercellular mitochondria exchange in
cancer drug resistance. Biochem J, 2018. 475(14): p. 2305-2328. https://doi.org/10.1042/BCJ20170712 PMid:30064989
- Koundouros, N. and G. Poulogiannis, Reprogramming of fatty acid metabolism in cancer. Br J Cancer, 2020. 122(1): p. 4-22. https://doi.org/10.1038/s41416-019-0650-z PMid:31819192 PMCid:PMC6964678
- Zhang,
H, Li H, Ho N, Li D, Li S. Scd1 plays a tumor-suppressive role in
survival of leukemia stem cells and the development of chronic myeloid
leukemia. Mol Cell Biol, 2012. 32(10): p. 1776-87. https://doi.org/10.1128/MCB.05672-11 PMid:22431519 PMCid:PMC3347408
- Scaglia,
N. and R.A. Igal, Stearoyl-CoA desaturase is involved in the control of
proliferation, anchorage-independent growth, and survival in human
transformed cells. J Biol Chem, 2005. 280(27): p. 25339-49.
https://doi.org/10.1074/jbc.M501159200 PMid:15851470
- Freeman
DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz
H, Rozengurt N, Cardiff RD, Liu X, Wu H. PTEN tumor suppressor
regulates p53 protein levels and activity through phosphatase-dependent
and -independent mechanisms. Cancer Cell, 2003. 3(2): p. 117-30. https://doi.org/10.1016/S1535-6108(03)00021-7 PMid:12620407
- Diaz-Blanco
E, Bruns I, Neumann F, Fischer JC, Graef T, Rosskopf M, Brors B,
Pechtel S, Bork S, Koch A, Baer A, Rohr UP, Kobbe G, von Haeseler A,
Gattermann N, Haas R, Kronenwett R. Molecular signature of CD34(+)
hematopoietic stem and progenitor cells of patients with CML in chronic
phase. Leukemia, 2007. 21(3): p. 494-504. https://doi.org/10.1038/sj.leu.2404549 PMid:17252012
- Baccarani,
M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, Apperley
J, Cervantes F, Cortes J, Deininger M, Gratwohl A, Guilhot F, Horowitz
M, Hughes T, Kantarjian H, Larson R, Niederwieser D, Silver R, Hehlmann
R. Evolving concepts in the management of chronic myeloid leukemia:
recommendations from an expert panel on behalf of the European
LeukemiaNet. Blood, 2006. 108(6): p. 1809-20. https://doi.org/10.1182/blood-2006-02-005686 PMid:16709930
- Haznedaroglu,
I.C., Monitoring the Response to Tyrosine Kinase Inhibitor (TKI)
Treatment in Chronic Myeloid Leukemia (CML). Mediterr J Hematol Infect
Dis, 2014. 6(1): p. e2014009. https://doi.org/10.4084/mjhid.2014.009 PMid:24455118 PMCid:PMC3894837
- Hekmatshoar
Y, Ozkan T, Altinok Gunes B, Bozkurt S, Karadag A, Karabay AZ,
Sunguroglu A. Characterization of imatinib-resistant K562 cell line
displaying resistance mechanisms. Cell Mol Biol (Noisy-le-grand), 2018.
64(6): p. 23-30. https://doi.org/10.14715/cmb/2018.64.6.5 PMid:29808796
- Ciftciler,
R. and I.C. Haznedaroglu, Tailored tyrosine kinase inhibitor (TKI)
treatment of chronic myeloid leukemia (CML) based on current evidence.
Eur Rev Med Pharmacol Sci, 2021. 25(24): p. 7787-7798.
- Fernandez,
L.P., M. Gomez de Cedron, and A. Ramirez de Molina, Alterations of
Lipid Metabolism in Cancer: Implications in Prognosis and Treatment.
Front Oncol, 2020. 10: p. 577420. https://doi.org/10.3389/fonc.2020.577420 PMid:33194695 PMCid:PMC7655926
- hen,
L., Ren J, Yang L, Li Y, Fu J, Li Y, Tian Y, Qiu F, Liu Z, Qiu Y.
Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer
by promoting ceramide synthesis. Sci Rep, 2016. 6: p. 19665. https://doi.org/10.1038/srep19665 PMid:26813308 PMCid:PMC4728559
- Tracz-Gaszewska,
Z. and P. Dobrzyn, Stearoyl-CoA Desaturase 1 as a Therapeutic Target
for the Treatment of Cancer. Cancers (Basel), 2019. 11(7). https://doi.org/10.3390/cancers11070948 PMid:31284458 PMCid:PMC6678606
- Huang,
J., Fan XX, He J, Pan H, Li RZ, Huang L, Jiang Z, Yao XJ, Liu L, Leung
EL, He JX.SCD1 is associated with tumor promotion, late stage and poor
survival in lung adenocarcinoma. Oncotarget, 2016. 7(26): p.
39970-39979. https://doi.org/10.18632/oncotarget.9461 PMid:27223066 PMCid:PMC5129985
- Presler,
M., Wojtczyk-Miaskowska A, Schlichtholz B, Kaluzny A, Matuszewski M,
Mika A, Sledzinski T, Swierczynski J. Increased expression of the gene
encoding stearoyl-CoA desaturase 1 in human bladder cancer. Mol Cell
Biochem, 2018. 447(1-2): p. 217-224. https://doi.org/10.1007/s11010-018-3306-z PMid:29396722 PMCid:PMC6133071
- Wang,
C., Shi M, Ji J, Cai Q, Zhao Q, Jiang J, Liu J, Zhang H, Zhu Z, Zhang
J.Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and
anti-ferroptosis of gastric cancer cells and predicts poor prognosis of
gastric cancer. Aging (Albany NY), 2020. 12(15): p. 15374-15391. https://doi.org/10.18632/aging.103598 PMid:32726752 PMCid:PMC7467382
- Southam
AD, Khanim FL, Hayden RE, Constantinou JK, Koczula KM, Michell RH,
Viant MR, Drayson MT, Bunce CM.Drug Redeployment to Kill Leukemia and
Lymphoma Cells by Disrupting SCD1-Mediated Synthesis of Monounsaturated
Fatty Acids. Cancer Res, 2015. 75(12): p. 2530-40. https://doi.org/10.1158/0008-5472.CAN-15-0202 PMid:25943877
- Morgan-Lappe
SE, Tucker LA, Huang X, Zhang Q, Sarthy AV, Zakula D, Vernetti L,
Schurdak M, Wang J, Fesik SW. Identification of Ras-related nuclear
protein, targeting protein for xenopus kinesin-like protein 2, and
stearoyl-CoA desaturase 1 as promising cancer targets from an
RNAi-based screen. Cancer Res, 2007. 67(9): p. 4390-8. https://doi.org/10.1158/0008-5472.CAN-06-4132 PMid:17483353
- Carter
BZ, Mak PY, Mu H, Zhou H, Mak DH, Schober W, Leverson JD, Zhang B,
Bhatia R, Huang X, Cortes J, Kantarjian H, Konopleva M, Andreeff M.
Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates
chronic myeloid leukemia stem cells. Sci Transl Med, 2016. 8(355): p.
355ra117. https://doi.org/10.1126/scitranslmed.aag1180
- Perini
GF, Ribeiro GN, Pinto Neto JV, Campos LT, Hamerschlak N. BCL-2 as
therapeutic target for hematological malignancies. J Hematol Oncol,
2018. 11(1): p. 65. https://doi.org/10.1186/s13045-018-0608-2 PMid:29747654 PMCid:PMC5946445
- Daflon-Yunes,
N., et al., Characterization of a multidrug-resistant chronic myeloid
leukemia cell line presenting multiple resistance mechanisms. Mol Cell
Biochem, 2013. 383(1-2): p. 123-35. https://doi.org/10.1007/s11010-013-1761-0 PMid:23877223
- Peng, C., Chen Y, Li D, Li S.Role of Pten in leukemia stem cells. Oncotarget, 2010. 1(2): p. 156-60. https://doi.org/10.18632/oncotarget.119 PMid:21297225 PMCid:PMC3157706
- Panuzzo
C, Crivellaro S, Carrà G, Guerrasio A, Saglio G, Morotti A. BCR-ABL
promotes PTEN downregulation in chronic myeloid leukemia. PLoS One,
2014. 9(10): p. e110682. https://doi.org/10.1371/journal.pone.0110682 PMid:25343485 PMCid:PMC4208795
[TOP]