Issanun Hunnuan1, Kleebsabai Sanpakit1, Ornsuda Lertbannaphong2 and Jassada Buaboonnam1.
1 Division
of Hematology and Oncology, Department of Pediatrics, Faculty of
Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand.
2
Division of Endocrinology, Department of Pediatrics, Faculty of
Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand.
Correspondence to:
Jassada Buaboonnam, MD. Associate Professor of Pediatrics. Division of
Hematology and Oncology, Department of Pediatrics, Faculty of Medicine
Siriraj Hospital, Mahidol University. 2 Wanglang Road, Bangkoknoi,
Bangkok 10700, Thailand. Tel: +66 2 419 5960; Fax: +66 2 411 3010
Email:
onco008@yahoo.com
Published: September 1, 2023
Received: May 10, 2023
Accepted: August 8, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023045 DOI
10.4084/MJHID.2023.045
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Hemoglobin H disease (HbH), a hemoglobinopathy resulting from abnormal
alpha globin genes, is classified into two categories: deletional HbH
(DHbH) and non-deletional HbH (NDHbH). The alpha-mutation genotypes
exhibit a range of clinical anemias, which differentially impact
patient growth.
Objectives: This retrospective study assessed the growth of HbH patients at Siriraj Hospital, Mahidol University.
Methods:
Patients diagnosed with HbH between January 2005 and April 2021 were
analyzed using growth standard scores of the Thai Society for Pediatric
Endocrinology (2022 version) and BMI-for-age Z scores of the World
Health Organization. Growth failure was defined as a patient’s height
for age exceeding two standard deviations below the mean.
Results: Of the 145 HbH patients, 75 (51.7%) had NDHbH, with --SEA/αCSα
being the most common genotype (70 patients; 93.3%). The mean baseline
hemoglobin level was significantly lower in NDHbH patients than in DHbH
patients (8.16 ± 0.93 g/dL vs. 9.51 ± 0.68 g/dL; P < 0.001). Splenomegaly and growth failure prevalences were higher in NDHbH patients (37.3% vs. 0%, with P < 0.001, and 22.7% vs. 8.6%, with P
= 0.020, respectively). Multivariable analysis revealed splenomegaly
> 3 cm was associated with growth failure (OR = 4.28; 95% CI,
1.19–15.39; P = 0.026).
Conclusions:
NDHbH patients exhibited lower hemoglobin levels and more pronounced
splenomegaly than DHbH patients. Growth failure can occur in both HbH
types but appears more prevalent in NDHbH. Close monitoring of growth
velocity is essential, and early treatment interventions may be
required to prevent growth failure.
|
Background
The
hemoglobin (Hb) protein is composed of two alpha globin chains (α
chains) and two beta-globin chains (β chains), arranged in a tetramer
of α2β2. Usually, each chromosome 16 contains two alpha genes,
composing four alpha genes per genome.[1] Natural
mutations, mainly deletion of the alpha globin gene, lead to alpha
thalassemia. Hemoglobin H disease (HbH), the most common form of alpha
thalassemia syndrome, results from compound heterozygosity of α0
thalassemia due to a loss of two linked alpha globin genes and either
single alpha gene deletion (deletional HbH; DHbH) or a non-deletional
mutation (non-deletional HbH; NDHbH) on the other alleles. Therefore,
HbH can be classified into two types, i.e., DHbH and NDHbH.[2,3]
The genetic mutations of HbH vary among ethnicities. For instance, the deletional mutations (--MED) and (-α20.5) are commonly found in the Mediterranean region, whereas (--SEA), (--FIL), (--THAI), (-α-3.7), and (-α-4.2)
are prevalent in Southeast Asia. The Hb Constant Spring (CS) variant
(α2 codon 142 TAA>CAA) is the most common NDHbH. Other
non-deletional types include Hb Quang Sze (α2 codon 125 CTG>CCG), Hb
Paksé (α2 codon 142 TAA>TAT), Hb Q Thailand (α2 codon 34
GAC>CAC), Hb Saun Dok (α2 codon 109 CTG>CGG), α2 codon 59
(GGC>GAC), α2 codon 0 Δ1bp (-T), α2 codon 30 Δ3bp (-GAG), and α2
codon 35 (TCC>CCC).[4-6]
The prevalence ratio of DHbH to NDHbH is varied. Although DHbH was found to be more prevalent in several studies,[3,7,8] NDHbH was more prevalent in some studies from Thailand.[9-11] In the United States, Hong Kong, and Canada, the majority of DHbH cases have genotypes of --SEA/-α-3.7 (55%), --SEA/-α-4.2 (12%), and --FIL/-α-3.7 (11%), while NDHbH is caused mainly by the --SEA/αCSα genotype (10%).[6] In Thailand, the genotypic distribution of HbH is --SEA/-α-3.7 (33.3%–57.5%) and --SEA/αCSα (53%–55%).[10,12]
The
severity of the disease is contingent upon the specific
alpha-thalassemia type involved, with NDHbH generally presenting
greater clinical severity than DHbH.[10,13]
Most patients with HbH have mild anemia; a few patients may require
transfusion ranging from occasional transfusion to regular transfusion.[10]
Some patients who occasionally have received blood transfusion support
may also develop complications, particularly during adolescence. These
complications may be delayed growth and puberty and reduced final
height,[14] mainly due to chronic anemia and gonadal dysfunction.[15]
In HbH patients, growth development during the first ten years of life is typically normal.[12]
However, some patients, especially those with severe anemia, may
experience abnormal growth during pre-adolescents. Moreover, NDHbH
patients may experience growth retardation at a young age.[6]
Although
abnormal growth can significantly impact patients with HbH, there is
currently a scarcity of studies investigating this topic. Therefore,
our study aimed to identify factors associated with growth retardation
and other relevant complications in HbH patients. The information
gathered from this study may help physicians improve treatment outcomes
for individuals with HbH.
Materials and Methods
This
retrospective study was conducted on patients aged 1 month to 18 years
diagnosed with HbH at the Department of Pediatrics, Siriraj Hospital,
Mahidol University, Thailand, between January 2005 and April 2021. All
included neonate patients previously presented with anemia or neonatal
jaundice and were subsequently diagnosed with HbH.
The data
collected by this study were the frequency of hemolytic crises, the
number of occasional transfusions since diagnosis, history of
splenectomy, and age of growth failure. The genotypes of alpha globin
mutations were also recorded. To assess patients’ health status,
hemoglobin level, red blood cell indices, reticulocyte count index, and
serum ferritin were measured at three consecutive follow-up visits
while the patients were not experiencing acute hemolytic episodes.
Serum
ferritin and vitamin D levels were also assessed. Vitamin D status was
evaluated according to the Thai Society for Pediatric Endocrinology’s
2023 guidelines. Serum levels of 25-OHD less than 12 ng/mL, 12 to 20
ng/mL, and more than 20 to 100 ng/mL were defined as indicating vitamin
D deficiency, insufficiency, and sufficiency, respectively.
Patients’
weight and height were collected at each clinic visit. Height was
measured in the morning by a trained nurse using a wall-mounted
stadiometer. Patients’ longitudinal growth record data were assessed
using the growth standard score established by the Thai Society for
Pediatric Endocrinology in 2022. Body mass index (BMI) was calculated
as BMI = weight (kg) ÷ height2
(meters). Growth failure was diagnosed by a decline in height-for-age
greater than two standard deviations from the mean during follow-up.
Patients
with periodic anemic symptoms or hemoglobin less than 8 g/dl received
occasional transfusions, whereas those with chronic severe anemia
received regular transfusions to maintain pre-transfusion hemoglobin
more than 9 g/dl. The transfusion and chelation protocol of our
institute was previously described.[16]
The Institutional Review Board authorized the study protocol (approval number 127/2565 [IRB1]).
Statistical analysis.
Statistical analyses were performed using IBM SPSS Statistics, version
20 (IBM Corp, Armonk, NY, USA). Mann–Whitney, chi-square, Fisher’s
exact, and independent t-tests were used as appropriate to assess the
association between patient characteristics and growth failure.
Logistic regression analysis was also conducted to identify significant
factors associated with growth failure. A probability (P) value < 0.05 was considered statistically significant.
Results
The
study included 145 patients, 75 (51.7%) with NDHbH and 70 patients with
DHbH (48.3%). Among patients with NDHbH, most genotypes were the
Southeast Asian (SEA) deletion, followed by the THAI deletion. The
Constant Spring (CS) variant was the most common (96%), followed by
Paksé (PS; 4%).
Most NDHbH patients were compound heterozygous for the SEA type and the Constant Spring variant (--SEA/α-3.7α).
In contrast, most DHbH patients were compound heterozygous for the SEA
type and the 3.7-kb deletion of the α globin gene (--SEA/-α-3.7).
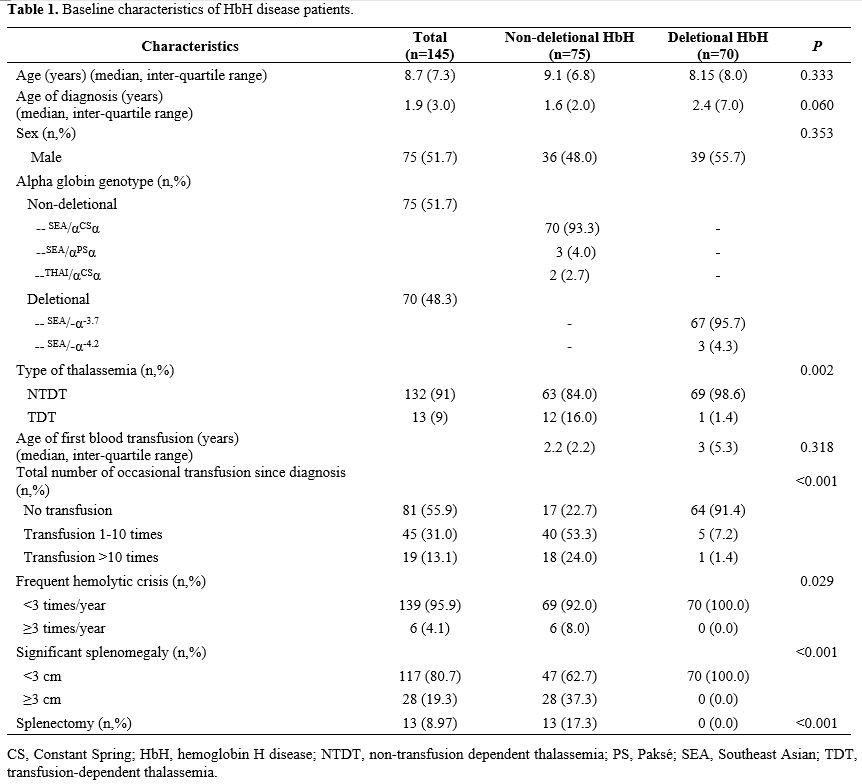
Table 1
presents the baseline clinical characteristics of the 145 HbH patients
included in this study. Of these patients, 23 (15.9%) had growth
failure, with a higher prevalence in NDHbH patients (17/75 patients;
22.7%) than in DHbH patients (6/70 patients; 8.6%). DHbH patients had
an earlier onset of growth failure, with a median age of onset of 1.6
(0.6–8.3) years compared to 7.4 (1–13.1) years for NDHbH patients.
 |
- Table
1. Baseline characteristics of HbH disease patients.
|
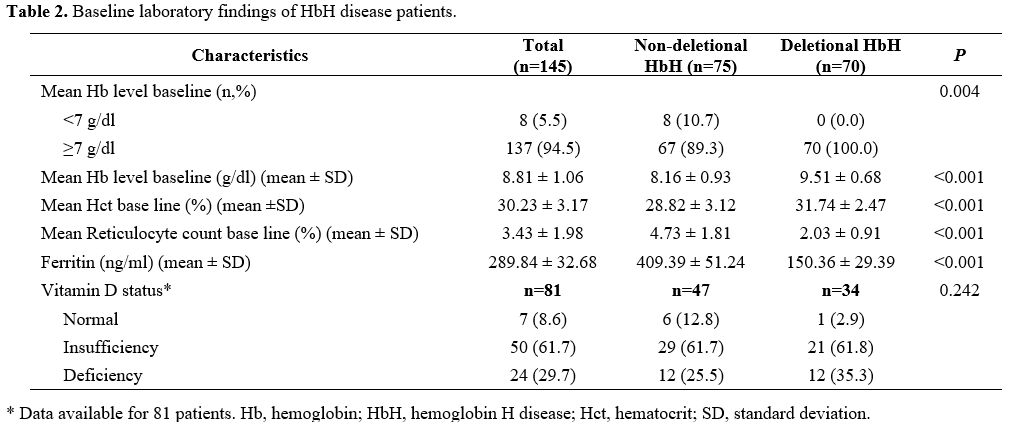
Table 2
displays the laboratory findings of HbH patients. Of the 81 patients
with available vitamin D data, 74 (91.3%) had low levels, comprising 50
(61.7%) with vitamin D insufficiency and 24 (29.7%) with vitamin D
deficiency. There was no significant difference in the prevalence of
low vitamin D between NDHbH and DHbH patients (P = 0.242).
 |
- Table 2. Baseline laboratory findings of HbH disease patients.
|
Table 3 demonstrates that NDHbH patients had lower weight for age, height for age, and weight for height than DHbH patients.
 |
- Table 3. A comparison of growth status of non-deletional HbH and deletional HbH patients.
|
Table 4
shows the correlation between clinical factors and growth failure. The
genotype most strongly associated with growth failure was --SEA/αCSα (69.6%), followed by --SEA/-α-3.7 (26.1%) and --SEA/αPSα
(4.3%). Multivariable analysis revealed that only splenomegaly was
significantly associated with growth failure (95% CI, 1.19–15.39; P = 0.026).
 |
- Table 4. Correlation of characteristics of the growth-failure and normal-height groups.
|
Discussion
NDHbH
was the most prevalent genotype in our study, accounting for 51.7% of
cases. This finding differs from other studies, but it is in agreement
with several studies conducted in tertiary care centers in Thailand.[9-11]
A possible explanation for this discrepancy is that our center is a
tertiary care referral center, with more severe cases, including those
with NDHbH, referred to our institution. Furthermore, this finding may
underscore that NDHbH has more severe anemia and may require treatment
intervention.
In Thailand, the --SEA/αCSα genotype variant was found to be the most prevalent among NDHbH patients, while the --SEA/-α-3.7 genotype variant was the most common among DHbH patients. This finding is consistent with other studies conducted in Thailand[11,12,15] and an investigation by Chao et al.[17] in Taiwan. However, our result differs from a survey by Shamoon et al.[18] in Iraq, which identified --MED/-α-3.7 as the most common genotype. These genotypic differences are likely related to ethnicity.
The
clinical severity of HbH disease can vary widely. This study found that
NDHbH was more severe than DHbH, consistent with other research.[10,12]
Specifically, NDHbH had lower mean hemoglobin levels and higher mean
reticulocyte counts at baseline than DHbH, as reported by Lal.[3]
Furthermore, in our cohort, NDHbH patients had a higher frequency of
hemolytic crises, a greater incidence of splenomegaly, and more
transfusions than DHbH patients. Approximately 9% of HbH patients in
this study underwent splenectomy, and all of them were NDHbH patients.
This finding is consistent with another study of Thai patients, which
reported a prevalence of splenectomy of 5%-8%.[10,12]
The role of vitamin D in bone health and mineralization is critical.[19] Adolescents commonly exhibit low levels of vitamin D.[20]
This study found a high prevalence of vitamin D insufficiency and
deficiency among alpha-thalassemia patients during clinical follow-up.
This result is consistent with other studies showing high rates of
vitamin D deficiency in thalassemia patients.[21,22]
This highlights the importance of monitoring vitamin D levels in this
patient population. Factors associated with decreased vitamin D levels
include avoidance of sun exposure, poor nutrition,[21] inadequate physical activity, and defective hydroxylation of vitamin D due to hepatic dysfunction.[23]
Health education, food fortification policies, and early detection
monitoring are necessary to mitigate the risk of vitamin D deficiency
and promote bone health and growth in thalassemia patients.[21]
Growth failure in thalassemia may be attributed to economic status[24]
and clinical factors such as the degree of chronic hypoxia, iron
overload, several micronutrient deficiencies, and parental height.[25,26] In this study, the prevalence of growth failure was 15%, consistent with other studies (13%-21%),[10,12] with the failure more pronounced among our patients with NDHbH.
Previous
research has shown that HbHCS is linked to more severe anemia and
growth failure that starts during infancy and early childhood,
requiring transfusions in children under the age of 6.[3] The --SEA/αCSα genotype has also been reported to have a significantly higher prevalence of growth failure.[20] Therefore, patients with HbHCS should be closely monitored for growth delay.
In our study, the BMI of thalassemia patients, both DHbH and NDHbH, was normal, as in another study.[20]
This finding highlights that monitoring growth in these patients should
rely on several parameters, not just BMI. In our cohort, splenomegaly
was also associated with growth failure, and patients with NDHbH or
splenomegaly should be closely monitored for growth. Early treatment
interventions such as regular transfusion and splenectomy may be
required to prevent growth failure.
There were some limitations to
our study. First, as growth is a dynamic process, factors such as
parental height, micronutrient levels, and another endocrine parameter
for evaluated growth may have confounded our results. These factors
could have acted as confounding variables. Another limitation of our
study is that it was retrospective, which meant that some data were
missing and could have introduced bias into our analyses. Finally,
since our center is a tertiary care referral center, the
generalizability of our findings to other centers may be limited.
Conclusions
Growth failure is common among patients with HbH,
particularly NDHbH. Close monitoring and multidisciplinary care are
essential to improve the quality of care for these patients.
Acknowledgments
The authors gratefully acknowledge Mr. David Park for language editorial assistance.
References
- Muncie HL, Jr., Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80(4):339-44.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167. https://doi.org/10.1016/S0140-6736(17)31822-6 PMid:28774421
- Lal
A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP.
Heterogeneity of hemoglobin H disease in childhood. N Engl J Med.
2011;364(8):710-8. https://doi.org/10.1056/NEJMoa1010174 PMid:21345100
- Bhat
VS, Dewan KK, Krishnaswamy PR. The diagnosis of α-Thalassaemia: A Case
of Hemoglobin H -α Deletion. Indian J Clin Biochem. 2010;25:435-40. https://doi.org/10.1007/s12291-010-0053-7 PMid:21966120 PMCid:PMC2994557
- Fucharoen
S, Viprakasit V. Hb H disease: clinical course and disease modifiers.
Hematology Am Soc Hematol Educ Program. 2009:26-34. https://doi.org/10.1182/asheducation-2009.1.26 PMid:20008179
- Chui DH, Fucharoen S, Chan V.Hemoglobin H disease:not necessarily a benign disorder.Blood. 2003;101(3):791-800. https://doi.org/10.1182/blood-2002-07-1975 PMid:12393486
- Chan
AY, So CC, Ma ES, Chan LC. A laboratory strategy for genotyping
haemoglobin H disease in the Chinese. J Clin Pathol.
2007;60:931-4. https://doi.org/10.1136/jcp.2006.042242 PMid:17018682 PMCid:PMC1994485
- Waye
JS, Eng B, Patterson M, Walker L, Carcao MD, Olivieri NF, et al.
Hemoglobin H (Hb H) disease in Canada: molecular diagnosis and review
of 116 cases. Am J Hematol. 2001;68:11-5.https://doi.org/10.1002/ajh.1142 PMid:11559931
- Boonsa,
S., Sanchaisuriya, K., Fucharoen, G., Wiangnon, S., Jetsrisuparb, A.,
& Fucharoen, S. (2004). The diverse molecular basis and
hematological features of Hb H and AEBart's diseases in Northeast
Thailand. Acta haematologica, 111(3), 149-154. https://doi.org/10.1159/000076523 PMid:15034236
- Charoenkwan
P, Taweephon R, Sae-Tung R, Thanarattanakorn P, Sanguansermsri T.
Molecular and clinical features of Hb H disease in northern Thailand.
Hemoglobin. 2005;29(2):133-40. https://doi.org/10.1081/HEM-58583 PMid:15921165
- Boonyawat
B, Photia A, Monsereenusorn C, Rujkijyanont P, Traivaree C. Molecular
characterization of Hb H and AEBart's diseases in Thai children:
Phramongkutklao hospital experiences. J Med Assoc Thai.
2017;100(2):167-74.
- Laosombat
V, Viprakasit V, Chotsampancharoen T, Wongchanchailert M, Khodchawan S,
Chinchang W, et al. Clinical features and molecular analysis in Thai
patients with HbH disease. Ann Hematol. 2009;88:1185-92. https://doi.org/10.1007/s00277-009-0743-5 PMid:19390853
- Songdej
D, Fucharoen S. Alpha-Thalassemia: Diversity of Clinical Phenotypes and
Update on the Treatment. Thalassemia Reports. 2022;12(4):157-172. https://doi.org/10.3390/thalassrep12040020
- Fung
EB, Harmatz PR, Lee PD, Milet M, Bellevue R, Jeng MR, et al. Increased
prevalence of iron-overload associated endocrinopathy in thalassaemia
versus sickle-cell disease. Br J Haematol. 2006;135(4):574-82. https://doi.org/10.1111/j.1365-2141.2006.06332.x PMid:17054676
- Pornprasert
S, Salaeh NA, Tookjai M, Punyamung M, Pongpunyayuen P, Treesuwan K.
Hematological analysis in Thai samples with deletional and
nondeletional HbH diseases. Lab Med. 2018;49(2):154-9. https://doi.org/10.1093/labmed/lmx068 PMid:29346671
- Buaboonnam
J, Takpradit C, Viprakasit V, Narkbunnam N, Vathana N, Phuakpet K, et
al. Long-term effectiveness, safety, and tolerability of twice-daily
dosing with deferasirox in children with transfusion-dependent
thalassemias unresponsive to standard once-daily dosing. Mediterr J
Hematol Infect Dis. 2021;13:e2021065. https://doi.org/10.4084/MJHID.2021.065 PMid:34804439 PMCid:PMC8577551
- Chao
YH, Wu KH, Wu HP, Liu SC, Peng CT, Lee MS. Clinical features and
molecular analysis of Hb H disease in Taiwan. Biomed Res Int.
2014;2014:271070. https://doi.org/10.1155/2014/271070 PMid:25309906 PMCid:PMC4163353
- Shamoon
RP, Yassin AK, Polus RK, Ali MD. Genotype-phenotype correlation of HbH
disease in northern Iraq. BMC Med Genet. 2020;21(1):203. https://doi.org/10.1186/s12881-020-01141-8 PMid:33059634 PMCid:PMC7559146
- Chatterjee
R, Bajoria R. Osteopenia-osteoporosis syndrome in patients with
thalassemia: understanding of type of bone disease and response to
treatment. Hemoglobin. 2009;33 Suppl 1:S136-8. https://doi.org/10.3109/03630260903347898 PMid:20001617
- Vogiatzi
MG, Macklin EA, Trachtenberg FL, Fung EB, Cheung AM, Vichinsky E, et
al. Differences in the prevalence of growth, endocrine and vitamin D
abnormalities among the various thalassaemia syndromes in North
America. Br J Haematol. 2009;146(5):546-56. https://doi.org/10.1111/j.1365-2141.2009.07793.x PMid:19604241 PMCid:PMC2798591
- Abdelmotaleb
GS, Behairy OG, El Azim KEA, El-Hassib DMA, Hemeda TM. Assessment of
serum vitamin D levels in Egyptian children with beta-thalassemia
major. Egypt Pediatric Association Gaz. 2021;69(1):20. https://doi.org/10.1186/s43054-021-00066-y
- Gombar
S, Parihar K, Choudhary M. Comparative study of serum ferritin and
vitamin D in thalassemia patients with healthy controls. Int J Res Med
Sci. 2018;6(2):693-5. https://doi.org/10.18203/2320-6012.ijrms20180322
- Fahim
FM, Saad K, Askar EA, Eldin EN, Thabet AF. Growth parameters and
vitamin D status in children with thalassemia major in upper Egypt. Int
J Hematol Oncol Stem Cell Res. 2013;7(4):10-4.
- Luo
HC, Luo QS, Huang FG, Wang CF, Wei YS. Impact of genotype on endocrinal
complications of children with alpha-thalassemia in China. Sci Rep.
2017;7(1):2948. https://doi.org/10.1038/s41598-017-03029-9 PMid:28592815 PMCid:PMC5462763
- Moayeri
H, Oloomi Z. Prevalence of growth and puberty failure with respect to
growth hormone and gonadotropins secretion in beta-thalassemia major.
Arch Iran Med. 2006;9(4):329-34.
- Tan
KA, Lum SH, Yahya A, Krishnan S, Jalaludin MY, Lee WS. Prevalence of
growth and endocrine disorders in Malaysian children with
transfusion-dependent thalassaemia. Singapore medical journal.
2019;60(6):303-8. https://doi.org/10.11622/smedj.2018155 PMid:30556093 PMCid:PMC6595058
[TOP]