Mohsen Nakhaie1,2,*, Zohreh-al-sadat Ghoreshi3,4,*, Mohammad Rezaei Zadeh Rukerd2,5, Hedyeh Askarpour3 and Nasir Arefinia3,6.
1 Student Research Committee, Kerman University of Medical Sciences, Kerman, Iran.
2
Gastroenterology and Hepatology Research Center, Institute of Basic and
Clinical Physiology Sciences, Kerman University of Medical Sciences,
Kerman, Iran.
3 School of Medicine, Jiroft University of Medical Sciences, Jiroft, Iran.
4 Student Research Committee Jiroft University of Medical Sciences, Jiroft, Iran.
5 Universal Scientific Education and Research Network (USERN), Tehran, Iran.
6 Bio Environmental Health Hazard Research Center, Jiroft University of Medical Sciences, Jiroft, Iran.
* The authors equally contributed to the work.
Correspondence to:
Nasir Arefinia, School of Medicine, Jiroft University of Medical
Sciences, Jiroft, Iran. Tel: +9834-42652781, FAX: +9871-42710780.
E-mail:
N.arefinia@jmu.ac.ir,
N.arefinia@gmail.com.
Published: November 1, 2023
Received: June 10, 2023
Accepted: October 12, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023059 DOI
10.4084/MJHID.2023.059
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Introduction:
Mutation in the genome of SARS-CoV-2 may play a role in immune evasion,
pathogenicity, and speed of its transmission. Our investigation aimed
to evaluate the mutations that exist in the NSP2.
Materials and Method:
RNA was extracted from nasopharyngeal swabs from 100 COVID-19 patients.
RT-PCR was performed on all samples using NSP2-specific primers.
Following gel electrophoresis, the bands were cut, purified, and
sequenced using the Sanger method. After sequencing, 90 sequences could
be used for further analysis. Bioinformatics analysis was conducted to
investigate the effect of mutations on protein structure, stability,
prediction of homology models, and phylogeny tree.
Results:
The patients' mean age was 51.08. The results revealed that 8 of the 17
NSP2 mutations (R207C, T224I, G262V, T265I, K337D, N348S, G392D, and
I431M) were missense. One deletion was also found in NSP2. Among NSP2
missense mutations studied, K337D and G392D increased structural
stability while the others decreased it. The homology-designed models
demonstrated that the homologies were comparable to the sequences of
the Wuhan-HU-1 virus.
Conclusion:
Our study suggested that the mutations K337D and G392D modulate the
stability of NSP2, and tracking viral evolution should be implemented
and vaccine development updated.
|
Introduction
Severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative
agent of the COVID-19 disease, is a highly contagious respiratory
pathogen.[1] COVID-19 initially emerged in Wuhan, China, in December 2019
and has spread rapidly worldwide, leading to a global pandemic.[2,3]
COVID-19 symptoms can manifest in a range of mild to severe forms,
including fever, cough, shortness of breath, fatigue, loss of taste or
smell, and more rarely, gastrointestinal bleeding, pneumonia, acute
respiratory distress syndrome (ARDS), and even death.[4-6] As of May 21,
2023, approximately 766 million cases of SARS-CoV-2 have been recorded,
along with 6.9 million reported deaths.[7,8] The genome sequencing
analysis of SARS-CoV-2 and SARS, conducted through the novel Coloured
Genomic Bootstrap (CGB) barcoding method, reveals that despite the
presence of genomic regions with mixed ancestry derived from horseshoe
bat viruses, a predominant over 97% of their genomes originates from
bats located in Yunnan, China.[9,10] However, another possibility is that
the virus could have infected other mammals and transferred to the
human population through the live animal market in Wuhan.[11,12]
The
SARS-CoV-2 genome has an approximate size of 29.8 kb and consists of 14
open reading frames (ORFs).[13] ORF1a and ORF1b are cleaved into 15
non-structural proteins (NSP) including NSP1 to NSP10, and NSP12 to
NSP16, by enzymatic function of NSP3 (papain-like protease) and NSP5
(chymotrypsin like protease).[14,15] The NSP2 disrupts host signaling
during infection by interacting with Prohibitin 1 (PHB1) and Prohibitin
2 (PHB2), which are components of the mitochondrial prohibitin
complex.[16,17] In addition, NSP2 plays a role in regulating calcium
homeostasis within cells, post-transcriptional suppression, and
associates with seven cellular proteins involved in vesicular
trafficking.[14,18,19] Finally, NSP2, by initiating translational
suppression, not only enables SARS-CoV-2 to evade the Interferon type I
response but also contributes to inflammation by activating NF-κB.[20,21]
SARS-CoV-2
mutations can significantly affect viral circulation, immune evasion,
and pathogenicity.[22] Frequent genetic variations in viruses can often
result in drug resistance or the evasion of effective vaccination
strategies.[23-25] As the virus spreads, it can help to survive in the
host cell by introducing mutations and altering the structure of its
proteins.[26] Viral polymerase, due to its limited or absent
proof-reading activity, can lead to frequent mutations.[27] One of the
key areas of concern has been the genetic variability in SARS-CoV-2
genomes. Recent research has identified sixty-one mutations, primarily
concentrated in NSP3, RNA-directed RNA polymerase (RdRp), and
Nucleocapsid proteins.[28] In-depth analysis of 59,541 SARS-CoV-2 genomic
sequences revealed significant mutations, with certain mutations such
as T85I and Q57H proving deleterious, while P323L exhibited a
stabilizing effect, offering insights into the virus's evolutionary
dynamics and potential impact on pathogenesis.[29] The rapid mutation
rate of SARS-CoV-2 has resulted in the emergence of new viral variants,
particularly in the spike protein's receptor-binding domain (RBD).
These mutations can potentially enhance viral transmission, increase
disease severity, and potentially reduce the effectiveness of immune
responses, monoclonal antibody treatments, and vaccines.[30]
Additionally, distinctive mutations in the SARS-CoV-2 ORF1ab
polyprotein (265 T→I, 4715 P→L, 5828 P→L, and 5865Y→C) serve as a
signature for the United States, altering nonstructural protein
structures and emphasizing their relevance in antiviral therapeutic
design.[31]
The absence of the NSP2 protein may compromise
viral replication, leading to a defect;[32,33] however, it is important
to note that viable viruses can still be produced despite its
removal.[34,35] Furthermore, genome sequencing of SARS-CoV-2 variants
during the COVID-19 pandemic revealed sites of positive selection in
NSP2, indicating the adaptation of humans as a specific host following
successful zoonotic co-transmission.[36] Importantly, the observed
inability to rescue this defect by expressing NSP2 from an alternative
genomic site highlights the indispensable role of the timing of NSP2
expression.[37,38] Despite the recent decrease in infected cases, the
possibility of further waves of infection is worrying. However, there
is also optimism that these potential outbreaks can be prevented or
reduced with careful measures and surveillance.[25]
The studies of
many researchers have been realized on the SARS-CoV-2 replication
mechanism, pathogenicity, and therapeutic strategies. The aim of this
study was to provide information about virus mutation, which has
important implications for disease progression and the development of
drugs or vaccines. In order to achieve this aim, the Open Reading Frame
1ab (ORF1ab) of SARS-CoV-2 was analyzed to evaluate the mutations
caused by selection pressure on the virus and their impact on viral
protein stability to infect human hosts, thereby promoting epidemic
spread.
Materials and Methods
Participants and Study Design.
100 COVID-19 patients were selected for the study from June to
September 2022 at Shafa Hospital, affiliated with Kerman University of
Medical Science, Kerman, Iran. The inclusion criteria for the study
involved patients who satisfied the diagnostic standard for COVID-19.[39]
The other inclusion criteria for study participants encompass the
following conditions: 1) purification kit (Qiagen GmbH, Germany). After
purification, it is sent to Baseline 1 (Gemini) company for sequencing
via the Applied Biosystems 3730xl DNA Analyzer. Of the total sequences
submitted, 90 Confirmation of SARS-CoV-2 infection through a throat
swab with a cycle threshold (Ct) value less than 24, 2) Manifestation
of clinical symptoms such as chest pain, cough with bloody or purulent
sputum, diarrhea, dehydration, vomiting, and dyspnea, 3) Absence of
reported symptoms associated with underlying medical conditions, and 4)
Non-receipt of any of the existing COVID-19 vaccinations. A structured
questionnaire was obtained for demographic information, medical
history, breath number, and temperature. Before participation, all
individuals provided written informed consent, and the Ethics Committee
of Kerman University of Medical Sciences approved the study
(IR.KMU.REC.1402.024).
Samples.
All patients underwent nasopharyngeal sample collection through the use
of a specific swab. Samples are transferred to viral transfer media
(VTM) and then translocated by a cool box.
Extraction of RNA.
RNA isolation kit (Product no: 11856022001, Roje) was utilized to
extract viral RNA from oropharynx/nasopharynx samples following the
manufacturer's instructions. The concentration and purity of the
extracted RNA were evaluated by measuring 1 μl of each sample with
NanoDrop™ 2000 (Thermo Scientific, USA). Also, RNA integrity was
assessed for its quality control by running 4 μl of the extracted RNA
along with 2 μl Loading Day on 2% agarose gel.
Sequencing of NSP2.
The extracted RNA samples were converted to complementary DNA (cDNA)
using cDNA synthesis kits (Yekta-Tajhiz, Iran) according to the
manufacturer’s instructions. The sequencing of NSP2 of the ORF1ab gene
was performed on 90 confirmed cases of COVID-19. The NCBI database and
AlleleID software were used to extract the sequence of NSP2 and the
design of specific primers for the target region, respectively.
Conventional PCR techniques with Eppendorf Mastercycler were used to
amplify the NSP2. PCR conditions included 1X PCR buffer, 300 - 400 ng
of template cDNA, 1 mM MgCl2, 100 mM deoxynucleotide triphosphates
(dNTPs), 10 pmol of each primer, and 0.5 U of Taq polymerase in a total
volume of 25 µL. The PCR thermal profile was 95°C for 5 min, followed
by 40 cycles of 95°C for 45 second, 59.4°C for 45 second, 72°C for 35
second, and a final extension for 5 min at 72°C. The primer sequence
and annealing temperatures used for NSP2 amplification have been shown
in Table 1.
 |
- Table 1. The NSP2-specific primer sequences utilized for RT-PCR.
|
For
band detection, the products of the PCR amplification were subjected to
electrophoresis on a 1.5% agarose gel using a 100 bp molecular weight
marker. The bands corresponding to the 970bp studied region were
located and excised from the gel for purification through the MinElute
electrophoresis band results were clear and perfect for further
analysis, including CLC6 and Clustal Omega for aligning and
supplementing to obtain the sequence. The sequences were
cross-referenced with existing databases to validate the results using
the BLAST online search tool at www.ncbi.nlm.nih.gov. Furthermore, the
sequences were compared to the reference strain Wuhan-Hu to identify
any related mutations by utilizing Clustal Omega.
Variations of nucleotide.
We conducted multiple sequence alignments to identify any nucleotide
variations by using Clustal Omega.[40] The reference genome was the
Whuhan-Hu-1 strain sequence, with GenBank accession number 045512.
Clustal Omega's MVIEW program was used to analyze the alignment file.[41]
Variations of amino acid.
Each protein's multiple sequence alignment was analyzed using MVIEW
after being aligned again with Clustal Omega. In addition, amino acid
variation was detected by comparing it to the reference strain protein.
The impacts of genetic mutations.
Different prediction tools were employed to examine missense mutations'
stability change and structural consequences. Specifically, I-mutant
was utilized to determine structure stability.[26] Furthermore, Mutpred2 was employed to predict the molecular consequences and functional impact of missense mutations.[42]
Molecular Dynamic Simulations. Molecular dynamic (MD) simulations were performed using GROMACS96 43a1 program with CHARMM27 force field.[43]
Each system was solvated with TIP3P water with a minimal distance of
1.0 nm between the solute and the wall of the dodecahedron box.
Ionization states were assigned to titratable residues corresponding to
the pH 7.0 condition. A proper amount of Na and Cl ions was added
instead of water molecules to imitate an ionic strength of 0.15 M. The
system was then energy minimized using the steepest descent algorithm
with an initial step size of 0.01 nm for a maximum force of 1000
kJ/mol/nm and a maximum of 50,000 steps. Then, a 100-ps-long
unconstrained equilibration MD simulation was done at a constant
temperature (300 K) and pressure using Berendsen and Parrinello–Rahman
coupling methods. Pressure coupling was performed using a reference
pressure of 1.0 bar and a time constant of 1.0 ps. Finally, a
50-ns-long production MD simulation was performed at a constant
temperature of 300 K, maintained by the v-rescale thermostat.
Phylogenetic analysis.
Phylogenetic and sequence analyses were conducted to investigate the
evolutionary relationships between different isolates. Of the 90
samples with clear and complete sequencing results, 39 were selected
for constructing the phylogenetic tree. The remaining cases were
excluded either due to the similarity in their mutations or the absence
of mutations in their sequences, which would hinder the construction of
the phylogenetic tree. Reference sequences from the Wuhan strain and
concerning variants, including B.1.1.617.2, B.1.1.7, B.1.1.529,
B.1.351, and P.1, were used for analysis and phylogenetic tree
construction. Clustering of sequences was performed with MEGA
11software, followed by inference of evolutionary history using the
Maximum Likelihood method with ~1000 bootstrap iterations.
Results
Demographic
data. Out of the total submitted sequences, 90 results for NSP2 having
clear and complete sequencing were included. The nucleic acid sequences
were obtained from 48 males and 42 females, and patients' mean ages
were 51.08.
The SARS-CoV-2 variant classification. The
SARS-CoV-2 variant classification refers to categorizing different
strains or variants of the SARS-CoV-2 virus based on specific genetic
mutations or changes in its genome. These variants are identified
through genomic sequencing and analysis, which helps understand the
spread, evolution, and potential impact of different viral lineages.
The classification typically involves assigning names or designations
to different variants based on their specific mutations, such as
Alpha/B.1.1.7, Beta/B.1.351, Gamma/P.1, Delta/B.1.1.617.2,
Omicron/B.1.1.529, and so on. The variant classification provides
important information for monitoring the virus's global spread
assessing its transmissibility, virulence, and potential impact on
diagnostics, therapeutics, and vaccines. It helps researchers, public
health authorities, and healthcare professionals to track and respond
to the emergence and prevalence of different SARS-CoV-2 variants.
Mutations
found in SARS-CoV-2 isolates from Kerman. All 90 Kerman isolates'
analysis showed 17 Single-nucleotide polymorphisms (SNP) in the NSP2. A
deletion was also identified for NSP2 isolate (Table 2). Out of 17
mutations, there were 8 missense mutations in positions 207, 224, 262,
265, 337, 348, 392, and 431, whereas the remaining mutations were
synonymous (Table 3).
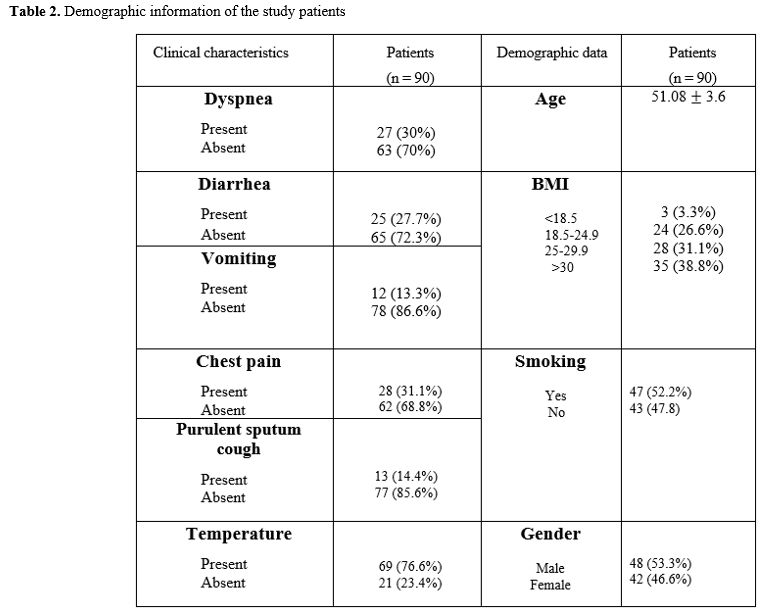 |
Table 2. Demographic information of the study patients. |
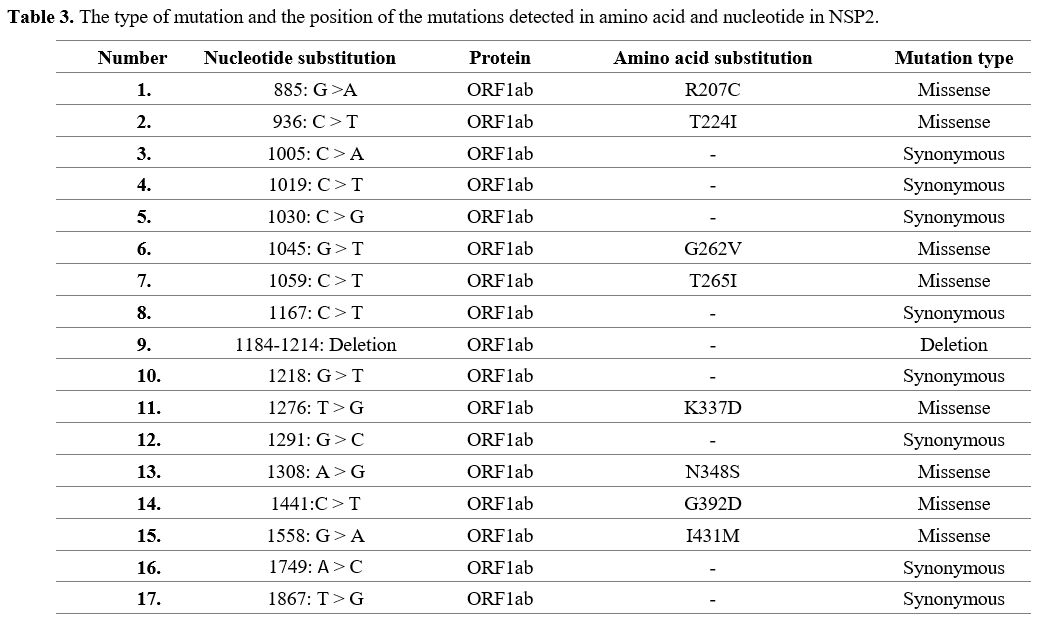 |
Table 3. The type of mutation and the position of the mutations detected in amino acid and nucleotide in NSP2.
|
Effects of various mutations
in NSP2. Six of the eight missense mutations identified in NSP2
indicated decreased structural stability. In addition, two remaining
mutations have an increase in stability of structural (Table 4).
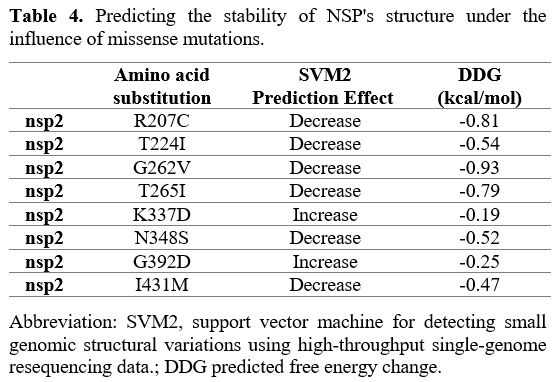 |
- Table 4. Predicting the stability of NSP's structure under the influence of missense mutations.
|
Root
mean square deviation. Root mean square deviation (RMSD) was also used
to investigate the stability of the structures further. It was assessed
during the 50-ns MD simulation runs for WT nsp2 and R207C, T224I,
G262V, T265I, K337D, N348S, G392D, I431M nsp2 systems. Unlike other
mutations, the RMSD in the K337D (Purple) and the G392D (Brown)
mutation were lower compared to the wild variant, indicating that these
mutations were more structurally stable (Figure 1).
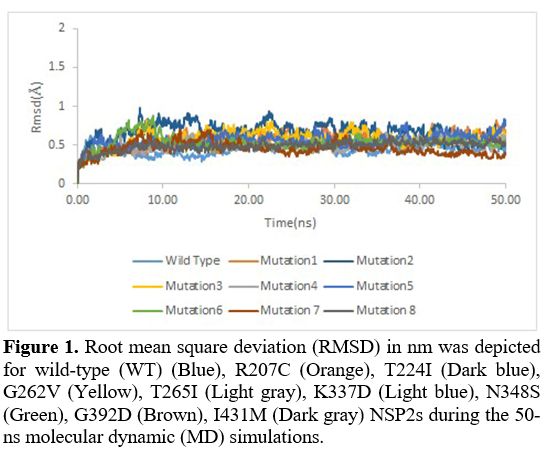 |
- Figure 1. Root mean
square deviation (RMSD) in nm was depicted for wild-type (WT) (Blue),
R207C (Orange), T224I (Dark blue), G262V (Yellow), T265I
- (Light gray),
K337D (Light blue), N348S (Green), G392D (Brown), I431M (Dark gray)
NSP2s during the 50-ns molecular dynamic (MD) simulations.
|
Anticipation
and verification of homology model predictions. Nine models were
produced for NSP2 using the PDB ID 7MSX as a template: the eight models
were for the Kerman isolate, and the remaining one was for the
reference strain. Mutant models 1 to 8 are designed for R207C, T224I,
G262V, T265I, K337D, N348S, G392D, and I431M mutations, respectively.
The reliability of these 8 models was assessed with validation
assessment scores, which were similar to the template (Table 5).
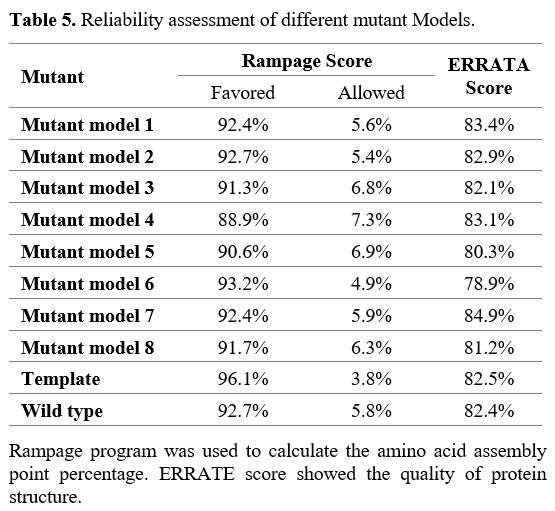 |
- Table 5. Reliability assessment of different mutant Models.
|
Phylogenetic
analysis. In this study, we compared the NSP2 sequencing results to
globally registered concern variants and provided detailed
specifications in Figure 2. According to our study, the predominant
SARS-CoV-2 viruses circulating were comparable to the Delta/B.1.617.2
variant (Figure 2). In addition, we found that the NSP2 variant of
concern (VOCs) prevalence with Pangolin Lineages Delta was 89.74% while
Omicron/B.1.1.529 accounted for 5.1% (Figure 2).
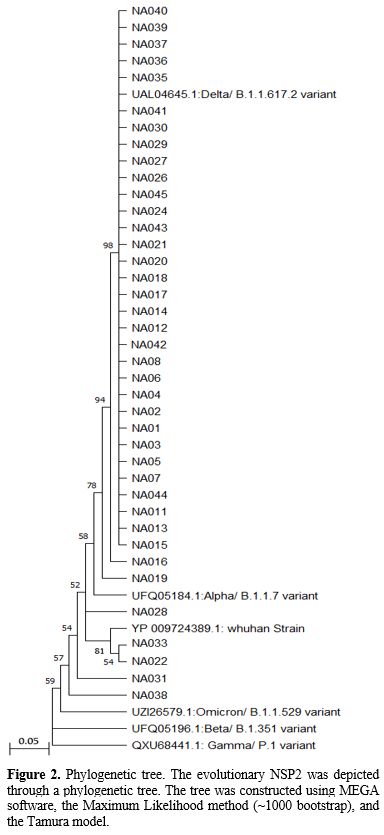 |
- Figure 2.. Phylogenetic
tree. The evolutionary NSP2 was depicted through a phylogenetic tree.
The tree was constructed using MEGA software, the Maximum
- Likelihood
method (~1000 bootstrap), and the Tamura model.
|
Discussion
The COVID-19 pandemic has emerged as a worldwide crisis.[24] Viral
genome mutations and subsequent modification of viral proteins are
common ways for viruses to escape from the immune system response and
survive within the host for extended periods.[34,37] Although cases of
the disease have decreased recently, there are still concerns about the
possibility of further waves of infection.[36,38] This study analyzed
the sequencing of SARS-CoV-2 in Kerman, Iran, and identified variations
that could offer insights into the virus's pathogenesis, genetic
diversity of NSP2, and the potential impact of mutations. A total of 17
mutations were detected; 8 mutations were related to missense mutation,
including R207C, T224I, G262V, T265I, K337D, N348S, G392D, I431M, and
N348S, and one mutation was related to deletion.
The NSP2
contains two functional clusters, one containing three proteins
involved in vesicle transport.[44] In our study, some mutations, such as
V1883T and R207C, occurred in this cluster, which may result in the
effective virus release from the endosome. The remaining cluster
comprises eight proteins associated with ribosome assembly and has the
potential to obstruct the transcription and translation process of
human mRNAs.[45] Other mutations in this cluster include T224I, G262V,
and T265I, which may alter the pathogenic pathway of SARS-CoV-2 by
interfering with proteins or cellular signaling. A prior investigation
has demonstrated that NSP2 may impact calcium homeostasis, and
alterations such as N348S and I431M mutations could potentially modify
its function. This modification is significant because apoptosis is a
crucial mechanism for host cell defense against SARS-CoV-2 infection.[46]
According to the identification of these mutations in our study, it is
possible that the variants caused by mutations can alter cell apoptosis
by altering calcium homeostasis. On the other hand, some RNA-binding
proteins, such as STAU2, play an anti-apoptotic role in DNA replication
and genome integrity maintenance.[47] The binding of SARS-CoV-2 NSP2
protein to STAU2 may inhibit its anti-apoptotic function,[48] and the
mutations in NSP2 may affect this interaction.
The NSP2 can
interact with some of the guanine nucleotide exchange factors such as
RAP1GDS1, and by changing the activity of some small GTPases, it might
aggravate or alleviate lung inflammation during SARS-2
infection,[14,49] which is consistent with the mutations identified in
our study. Many organelles, including endosomes, lysosomes, and
exosomes, have protein complexes such as V-ATPase in their membranes,
which transfer protons to the organelle to maintain the acidic
environment. During SARS-CoV-2 infection, researchers have demonstrated
that NSP2 interacts with some subunits of V-ATPase and is involved in
transporting substances such as Ca2+.[50] Therefore, some mutations
that occurred in NSP2 can change the interaction between it and
V-ATPase and may participate in induced during endocytosis.
In
addition, NSP2 could be considered one of the targets of laboratory
diagnosis for SARS-CoV-2 by rapid and real-time reverse
transcriptase-polymerase chain reaction (rRT-PCR). In their study, Yip
et al. identified a 154 nucleotide fragment as a conserved region for
detecting SARS-CoV-2.[51] Therefore, any mutation at the 1867 position
in our study could potentially fail to identify SARS-CoV-2 patients.
Continuously monitoring mutations will be crucial to track the virus's
spread among individuals and across different regions.
Another
mutation was responsible for deletion. Large deletion mutations can
cause a defect in the production and function of the desired protein.
In this regard, some scientists have suggested that large deletions in
OFR1ab may not encode the target protein.[52] Large deletion mutations
have also been found in the studies of other scientists; for instance,
an 80-nucleotide deletion in ORF7a was also reported in a study
conducted in Arizona.[53]
In concordance with our study,
Banerjee et al. identified a prevalent mutation, specifically T265I,
within the NSP2 gene. Their research spanned 31 different states across
the United States, involving the examination of 867 complete protein
sequences of ORF1ab. Notably, they found that among the genes
comprising the ORF1ab region, which constitutes approximately
two-thirds of the SARS-CoV-2 genome, the T265I mutation exhibited the
highest incidence, accounting for around 50%. These findings are
consistent with our investigation, as the NSP gene is known to
influence mitochondrial function, manage cellular stress, and modulate
host cell survival signaling pathways by interacting with PHB and PHB2
proteins within the host organism. Consequently, the T265I mutation in
this gene may confer advantages to the virus in these processes.[31] In
alignment with the findings of Koyama et al., our study has also
highlighted the significance of the G392D mutation in the context of
SARS-CoV-2 evolution and its potential impact on viral stability.
Koyama et al.'s comprehensive analysis of over 10,000 SARS-CoV-2
genomes from diverse geographical regions revealed a spectrum of
genetic variants, including the G392D mutation.[54]
Conclusions
Our
study delves into the intricate realm of SARS-CoV-2 evolution, shedding
light on crucial aspects contributing to its adaptability and potential
impact on public health. Notably, we have elucidated a spectrum of
variations within NSP2, uncovering a nuanced narrative of viral
stability. While some genetic variations may undermine NSP2 stability,
intriguingly, others, such as K337D and G392D, appear to bolster it.
This dualistic insight into NSP2's stability diversification adds a
novel layer to our understanding of the virus's adaptive mechanisms.
Our work also extends beyond the realm of basic research. The homology
models we have meticulously designed to elucidate the structural
consequences of NSP2 mutations provide a valuable resource for future
studies aiming to decipher the functional implications of these genetic
changes. Importantly, these models align with the Wuhan strain and
reveal a striking resemblance to the Delta variant, underscoring the
relevance of our findings in the context of contemporary viral
evolution.
Furthermore, our findings carry significant
implications for public health. We emphasize the pressing need for
continuous surveillance of genomic variations within SARS-CoV-2,
especially in the face of emerging variants like Delta, to inform the
development of effective treatment strategies and updated vaccines. In
conclusion, our research offers a comprehensive and nuanced perspective
on SARS-CoV-2 evolution, accentuating our unique insights into NSP2
variations, their structural implications, and their relevance in
contemporary viral landscapes. This multi-faceted contribution advances
our understanding of the virus and provides a foundation for future
investigations to combat the ongoing global health challenge.
Acknowledgments.
The authors thank the Research Consultation Center for improving the article's writing.
Funding
This
work was supported by Student Research Committee, Kerman University of
Medical Sciences, Kerman, Iran (Grant numbers 401000085). Author Mohsen
Nakhaie has received research support from the Kerman University of
Medical Sciences.
Author Contributions
NA,
ZG designed the study, and NA Analyzed the data. NA and MN Contributed
new methods or models. NA and ZG wrote the paper. MRZR, NA, and MN
reviewed and finalized the manuscript. All authors read and approved
the final manuscript.
Ethics approval
This
study was performed in line with the principles of the Declaration of
Helsinki. Approval was granted by the Ethics Committee of Kerman
University of Medical Science (IR.KMU.REC.1402.024).
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publish.
The authors affirm that human research participants provided informed consent for publication.
References
- Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus
disease-2019 (COVID-19): The epidemic and the challenges. Int J
Antimicrob Agents. 2020 Mar;55(3):105924. https://doi.org/10.1016/j.ijantimicag.2020.105924 PMid:32081636 PMCid:PMC7127800
- Shi
J, Chen F, Chen S, Ling HQ. COVID-19 over the last three years in
China, what we've learned. Front Public Health. 11:1209343. https://doi.org/10.3389/fpubh.2023.1209343 PMid:37522001 PMCid:PMC10374005
- Listings of WHO's response to COVID-19 [Internet]. [cited 2023 Sep 29]. Available from: https://www.who.int/news/item/29-06-2020-covidtimeline
- Arefinia
N, Ghoreshi Z al S, Alipour AH, Reza Molaei H, Samie M, Sarvari J.
Gastrointestinal Manifestations in Patients Infected with SARS-CoV-2.
Iran J Med Microbiol. 2022 Jul 10;16(4):271-81. https://doi.org/10.30699/ijmm.16.4.271
- Shafieipour
S, Mohammadi E, Rukerd MRZ, Momenai R, Lashkarizadeh MM, Zahedi MJ, et
al. Gastrointestinal Bleeding: Prevalence, Etiology, and Outcomes in
COVID-19 Inpatients. GOVARESH. 2023 May 24;28(1):30-5.
- COVID-19 symptoms and severity [Internet]. [cited 2023 Sep 29]. Available from: https://www.who.int/westernpacific/emergencies/covid-19/information/asymptomatic-covid-19
- Weekly epidemiological update on COVID-19 - 25 May 2023 [Internet]. [cited 2023 Jul 14]. Available from: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---25-may-2023
- St
Clair LA, Chan LLY, Boretsky A, Lin B, Spedding M, Perera R.
High-throughput SARS-CoV-2 antiviral testing method using the Celigo
Image Cytometer. J Fluoresc. 2023;1-10. https://doi.org/10.1007/s10895-023-03289-x PMid:37310590 PMCid:PMC10261830
- Wang
LF, Eaton BT. Bats, civets and the emergence of SARS. Wildl Emerg
Zoonotic Dis Biol Circumst Consequences Cross-Species Transm.
2007;325-44. https://doi.org/10.1007/978-3-540-70962-6_13 PMid:17848070 PMCid:PMC7120088
- Hassanin
A, Rambaud O. Retracing Phylogenetic, Host and Geographic Origins of
Coronaviruses with Coloured Genomic Bootstrap Barcodes: SARS-CoV and
SARS-CoV-2 as Case Studies. Viruses. 2023;15(2):406. https://doi.org/10.3390/v15020406 PMid:36851620 PMCid:PMC9961909
- Zhang
N, Wang L, Deng X, Liang R, Su M, He C, et al. Recent advances in the
detection of respiratory virus infection in humans. J Med Virol.
2020;92(4):408-17. https://doi.org/10.1002/jmv.25674 PMid:31944312 PMCid:PMC7166954
- Brüssow
H. Viral infections at the animal-human interface-Learning lessons from
the SARS‐CoV‐2 pandemic. Microb Biotechnol. 2023; https://doi.org/10.1111/1751-7915.14269 PMid:37338856 PMCid:PMC10281366
- Zhang
Y, Huang Z, Zhu J, Li C, Fang Z, Chen K, et al. An updated review of
SARS‐CoV‐2 detection methods in the context of a novel coronavirus
pandemic. Bioeng Transl Med. 2023;8(1):e10356. https://doi.org/10.1002/btm2.10356 PMid:35942232 PMCid:PMC9349698
- Zheng
YX, Wang L, Kong WS, Chen H, Wang XN, Meng Q, et al. Nsp2 has the
potential to be a drug target revealed by global identification of
SARS-CoV-2 Nsp2-interacting proteins. Acta Biochim Biophys Sin. 2021
Aug 31;53(9):1134-41.https://doi.org/10.1093/abbs/gmab088 PMid:34159380
- Agrawal
PK, Agrawal C, Blunden G. Antiviral and Possible Prophylactic
Significance of Myricetin for COVID-19. Nat Prod Commun.
2023;18(4):1934578X231166283. https://doi.org/10.1177/1934578X231166283
- Cornillez-Ty
CT, Liao L, Yates JR 3rd, Kuhn P, Buchmeier MJ. Severe acute
respiratory syndrome coronavirus nonstructural protein 2 interacts with
a host protein complex involved in mitochondrial biogenesis and
intracellular signaling. J Virol. 2009 Oct;83(19):10314-8. https://doi.org/10.1128/JVI.00842-09 PMid:19640993 PMCid:PMC2748024
- Kabekkodu
SP, Chakrabarty S, Jayaram P, Mallya S, Thangaraj K, Singh KK, et al.
Severe acute respiratory syndrome coronaviruses contributing to
mitochondrial dysfunction: Implications for post-COVID complications.
Mitochondrion. 2023;69:43-56. https://doi.org/10.1016/j.mito.2023.01.005 PMid:36690315 PMCid:PMC9854144
- Davies
JP, Almasy KM, McDonald EF, Plate L. Comparative multiplexed
interactomics of SARS-CoV-2 and homologous coronavirus non-structural
proteins identifies unique and shared host-cell dependencies. bioRxiv :
the preprint server for biology. United States; 2020. https://doi.org/10.1101/2020.07.13.201517
- Senthilazhagan
K, Sakthimani S, Kallanja D, Venkataraman S. SARS-CoV-2: analysis of
the effects of mutations in non-structural proteins. Arch Virol. 2023
Jun 21;168(7):186. https://doi.org/10.1007/s00705-023-05818-2 PMid:37344726
- Xu
Z, Choi JH, Dai DL, Luo J, Ladak RJ, Li Q, et al. SARS-CoV-2 impairs
interferon production via NSP2-induced repression of mRNA translation.
Proc Natl Acad Sci U S A. 2022 Aug 9;119(32):e2204539119. https://doi.org/10.1073/pnas.2204539119 PMid:35878012 PMCid:PMC9371684
- Lacasse
É, Gudimard L, Dubuc I, Gravel A, Allaeys I, Boilard É, et al.
SARS-CoV-2 Nsp2 Contributes to Inflammation by Activating NF-κB.
Viruses. 2023 Jan 24;15(2):334. https://doi.org/10.3390/v15020334 PMid:36851549 PMCid:PMC9964531
- Sun
C, Xie C, Bu GL, Zhong LY, Zeng MS. Molecular characteristics, immune
evasion, and impact of SARS-CoV-2 variants. Signal Transduct Target
Ther. 2022 Jun 28;7:202. https://doi.org/10.1038/s41392-022-01039-2 PMid:35764603 PMCid:PMC9240077
- Delshad
M, Sanaei MJ, Pourbagheri-Sigaroodi A, Bashash D. Host genetic
diversity and genetic variations of SARS-CoV-2 in COVID-19 pathogenesis
and the effectiveness of vaccination. Int Immunopharmacol. 2022;109128.
https://doi.org/10.1016/j.intimp.2022.109128 PMid:35963158 PMCid:PMC9359488
- Yuen
KS, Ye ZW, Fung SY, Chan CP, Jin DY. SARS-CoV-2 and COVID-19: The most
important research questions. Cell Biosci. 2020;10(1):1-5. https://doi.org/10.1186/s13578-020-00404-4 PMid:32190290 PMCid:PMC7074995
- Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682. https://doi.org/10.1016/j.genrep.2020.100682 PMid:32300673 PMCid:PMC7161481
- Capriotti
E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes
upon mutation from the protein sequence or structure. Nucleic Acids
Res. 2005 Jul 1;33(suppl_2):W306-10. https://doi.org/10.1093/nar/gki375 PMid:15980478 PMCid:PMC1160136
- Herman
C, Bradley C, Gordon A, Wang C, Cooke M, Kohrn B, et al. RNA polymerase
inaccuracy underlies SARS-CoV-2 variants and vaccine heterogeneity.
Research square. United States; 2022. https://doi.org/10.21203/rs.3.rs-1690086/v1
- Periwal
N, Rathod SB, Pal R, Sharma P, Nebhnani L, Barnwal RP, et al. In silico
characterization of mutations circulating in SARS-CoV-2 structural
proteins. J Biomol Struct Dyn. 2022 Nov;40(18):8216-31. https://doi.org/10.1080/07391102.2021.1908170 PMid:33797336 PMCid:PMC8043164
- Periwal
N, Rathod SB, Sarma S, Johar GS, Jain A, Barnwal RP, et al. Time Series
Analysis of SARS-CoV-2 Genomes and Correlations among Highly Prevalent
Mutations. Microbiol Spectr. 2022 Oct 26;10(5):e0121922. https://doi.org/10.1128/spectrum.01219-22 PMid:36069583 PMCid:PMC9603882
- Flores-Vega
VR, Monroy-Molina JV, Jiménez-Hernández LE, Torres AG, Santos-Preciado
JI, Rosales-Reyes R. SARS-CoV-2: Evolution and Emergence of New Viral
Variants. Viruses. 2022 Mar 22;14(4):653. https://doi.org/10.3390/v14040653 PMid:35458383 PMCid:PMC9025907
- Banerjee
S, Seal S, Dey R, Mondal KKr, Bhattacharjee P. Mutational spectra of
SARS‐CoV‐2 orf1ab polyprotein and signature mutations in the United
States of America. J Med Virol. 2021 Mar;93(3):1428-35. https://doi.org/10.1002/jmv.26417 PMid:32779784 PMCid:PMC7436414
- Meshram
CD, Lukash T, Phillips A, Akhrymuk I, Frolova EI, Frolov I. Lack of
nsP2-specific nuclear functions attenuates chikungunya virus
replication both in vitro and in vivo. Virology. 2019 Aug;534:14-24. https://doi.org/10.1016/j.virol.2019.05.016 PMid:31163352 PMCid:PMC7204530
- Cherkashchenko
L, Rausalu K, Basu S, Alphey L, Merits A. Expression of Alphavirus
Nonstructural Protein 2 (nsP2) in Mosquito Cells Inhibits Viral RNA
Replication in Both a Protease Activity-Dependent and -Independent
Manner. Viruses. 2022 Jun 17;14(6):1327. https://doi.org/10.3390/v14061327 PMid:35746799 PMCid:PMC9228716
- Graham
RL, Sims AC, Brockway SM, Baric RS, Denison MR. The nsp2 replicase
proteins of murine hepatitis virus and severe acute respiratory
syndrome coronavirus are dispensable for viral replication. J Virol.
2005 Nov;79(21):13399-411. https://doi.org/10.1128/JVI.79.21.13399-13411.2005 PMid:16227261 PMCid:PMC1262610
- Zhang
L, Shen M, Ma X, Su S, Gong W, Wang J, et al. What is required to
prevent a second major outbreak of SARS-CoV-2 upon lifting quarantine
in Wuhan City, China. The Innovation. 2020;1(1):100006. https://doi.org/10.1016/j.xinn.2020.04.006 PMid:33458717 PMCid:PMC7237941
- Zhao
J, Sun J, He WT, Ji X, Gao Q, Zhai X, et al. Snapshot of the evolution
and mutation patterns of SARS-CoV-2. bioRxiv. 2020 Jan
1;2020.07.04.187435. https://doi.org/10.1101/2020.07.04.187435
- Gadlage
MJ, Graham RL, Denison MR. Murine coronaviruses encoding nsp2 at
different genomic loci have altered replication, protein expression,
and localization. J Virol. 2008 Dec;82(23):11964-9. https://doi.org/10.1128/JVI.01126-07 PMid:18815297 PMCid:PMC2583644
- Hodcroft
EB, Domman DB, Oguntuyo K, Snyder DJ, Diest M Van, Densmore KH, et al.
Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein
variants affecting amino acid position 677. medRxiv. 2021 Jan
1;2021.02.12.21251658. https://doi.org/10.1101/2021.02.12.21251658
- Emergency F, Only U, Only R. Real-Time RT-PCR Diagnostic Panel For Emergency Use Only. 2021.
- Madeira
F, Park Y mi, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The
EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids
Res. 2019 Jul 2;47(W1):W636-41. https://doi.org/10.1093/nar/gkz268 PMid:30976793 PMCid:PMC6602479
- Malik
JA, Ahmed S, Mir A, Shinde M, Bender O, Alshammari F, et al. The
SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to
new challenges. J Infect Public Health. 2022;15(2):228-40. https://doi.org/10.1016/j.jiph.2021.12.014 PMid:35042059 PMCid:PMC8730674
- Pejaver
V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam HJ, et al.
Inferring the molecular and phenotypic impact of amino acid variants
with MutPred2. Nat Commun. 2020;11(1):1-13. https://doi.org/10.1038/s41467-020-19669-x PMid:33219223 PMCid:PMC7680112
- Abraham
MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High
performance molecular simulations through multi-level parallelism from
laptops to supercomputers. SoftwareX. 2015 Sep 1;1-2:19-25. https://doi.org/10.1016/j.softx.2015.06.001
- Gordon
DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A
SARS-CoV-2 protein interaction map reveals targets for drug
repurposing. Nature. 2020;583(7816):459-68. https://doi.org/10.1038/s41586-020-2286-9 PMid:32353859 PMCid:PMC7431030
- Banerjee
AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2
Disrupts Splicing, Translation, and Protein Trafficking to Suppress
Host Defenses. Cell. 2020 Nov;183(5):1325-1339.e21. https://doi.org/10.1016/j.cell.2020.10.004 PMid:33080218 PMCid:PMC7543886
- Daniloski
Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, et al.
Identification of Required Host Factors for SARS-CoV-2 Infection in
Human Cells. Cell. 2021 Jan;184(1):92-105.e16. https://doi.org/10.1016/j.cell.2020.10.030 PMid:33147445 PMCid:PMC7584921
- Zhang
X, Trépanier V, Beaujois R, Viranaicken W, Drobetsky E, DesGroseillers
L. The downregulation of the RNA-binding protein Staufen2 in response
to DNA damage promotes apoptosis. Nucleic Acids Res. 2016
May;44(8):3695-712. https://doi.org/10.1093/nar/gkw057 PMid:26843428 PMCid:PMC4856980
- Oberdoerffer
S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45
alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL.
Science. 2008 Aug;321(5889):686-91. https://doi.org/10.1126/science.1157610 PMid:18669861 PMCid:PMC2791692
- Hamel
B, Monaghan-Benson E, Rojas RJ, Temple BRS, Marston DJ, Burridge K, et
al. SmgGDS is a guanine nucleotide exchange factor that specifically
activates RhoA and RhoC. J Biol Chem. 2011;286(14):12141-8. https://doi.org/10.1074/jbc.M110.191122 PMid:21242305 PMCid:PMC3069418
- Zhao
W, Gao X, Qiu S, Gao B, Gao S, Zhang X, et al. A subunit of V-ATPases,
ATP6V1B2, underlies the pathology of intellectual disability.
EBioMedicine. 2019 Jul;45:408-21. https://doi.org/10.1016/j.ebiom.2019.06.035 PMid:31257146 PMCid:PMC6642280
- Yip
CCY, Ho CC, Chan JFW, To KKW, Chan HSY, Wong SCY, et al. Development of
a Novel, Genome Subtraction-Derived, SARS-CoV-2-Specific COVID-19-nsp2
Real-Time RT-PCR Assay and Its Evaluation Using Clinical Specimens. Int
J Mol Sci. 2020 Apr;21(7). https://doi.org/10.3390/ijms21072574 PMid:32276333 PMCid:PMC7177594
- Alam
S, Mahfujur M, Morshed N. Since January 2020 Elsevier has created a
COVID-19 resource centre with free information in English and Mandarin
on the novel coronavirus COVID-19. The COVID-19 resource centre is
hosted on Elsevier Connect, the company's public news and information.
2020;(January).
- Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol. 2020;11:1800. https://doi.org/10.3389/fmicb.2020.01800 PMid:32793182 PMCid:PMC7387429
- Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. 2020 Jul 1;98(7):495-504. https://doi.org/10.2471/BLT.20.253591 PMid:32742035 PMCid:PMC7375210