Giulia
Falconi1, E Galossi1, H
Hajrullaj1, E Fabiani1,2
and MT Voso1.
1 Department
of Biomedicine and Prevention, University of Rome Tor Vergata, Rome,
Italy
2 UniCamillus-Saint Camillus International
University of Health Sciences, Rome, Italy.
Correspondence to: Prof.ssa
Maria Teresa Voso. Dipartimento di Biomedicina e Prevenzione,
Universita’ di Roma Tor Vergata Via Montpellier,1, 00133 Roma. E-mail:
voso@med.uniroma2.it
Published: September 1, 2023
Received: July 10, 2023
Accepted: August 14, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023055 DOI
10.4084/MJHID.2023.055
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Therapy-related
myeloid neoplasms (t-MN) are a late complication of cytotoxic therapy
(CT) used in the treatment of both malignant and non-malignant
diseases. Historically, t-MN has been considered to be a direct
consequence of DNA damage induced in normal hematopoietic stem or
progenitor cells (HSPC) by CT. However, we now know that
treatment-induced mutations in HSC are not the only players involved in
t-MN development but additional factors may contribute to the onset of
t-MN.
One of the known drivers involved in this field is the bone marrow
microenvironment (BMM) and, in particular, bone marrow mesenchymal stem
cells (BM-MSC), whose role in t-MN pathogenesis is the topic of this
mini-review.
BM-MSCs, physiologically, support HSC maintenance, self-renewal, and
differentiation through hematopoietic–stromal interactions and the
production of cytokines. In addition, BM-MSCs maintain the stability of
the BM immune microenvironment and reduce the damage caused to HSC by
stress stimuli.
In the t-MN context, chemo/radio-therapy may induce damage to the
BM-MSC and likewise alter BM-MSC functions by promoting
pro-inflammatory response, clonal selection and/or the production of
factors that may favor malignant hematopoiesis.
Over the last decade, it has been shown that BM-MSC isolated from
patients with de novo and therapy-related MN exhibit decreased
proliferative and clonogenic capacity, altered morphology, increased
senescence, defective osteogenic differentiation potential, impaired
immune-regulatory properties, and reduced ability to support HSC growth
and differentiation, as compared to normal BM-MSC.
Although the understanding of the genetic and gene expression profile
associated with ex vivo-expanded t-MN-MSCs remains limited and
debatable, its potential role in prognostic and therapeutic terms is
acting as a flywheel of attraction of many researchers.
|
Introduction
Therapy-related
myeloid neoplasms (t-MN), or MN post cytotoxic therapy (MN-pCT)
include, according to the 2022 WHO classification[1] and its previous editions,[2-3]
therapy-related acute myeloid leukemia (AML), myelodysplastic syndromes
(MDS), and myelodysplastic/myeloproliferative neoplasms (MDS/MPN).
t-MNs
are a late complication of cytotoxic agents (chemo and/or radiation
therapy) used in the treatment of both malignant (solid or
hematological) and non-malignant (mostly autoimmune) diseases.
They
are an emerging problem of our aging society, where the newer
therapeutic drugs and ameliorated cancer management protocols have
improved the life expectancy of cancer patients in the last decades.[4] This results in an increase in patient number at risk of developing this late treatment-related complication,[5] characterized by poor prognosis (5-year overall survival <10%)[6]
and refractoriness to current standard treatment strategies, still
remaining an unmet clinical need of cancer survivorship programs.[7]
t-MN accounts for approximately 10–20% of newly diagnosed cases of AML or MDS and can occur at any age.
Historically,
t-MN has been considered to be a direct consequence of DNA damage
induced in normal hematopoietic stem or progenitor cells (HSPC) by CT.
However, in recent years advances in deep sequencing techniques have
faltered this historical theory and have given way to a multi-hit model
of t-MN where both intrinsic and extrinsic factors contribute to its
development.[8]
According to this multi-step
pathogenesis, patient-related factors, including age, type, and
treatment of primary disease, in the presence of germ-line variants,
together with acquired factors, such as clonal hematopoiesis of
indeterminate potential (CHIP) and inflammation, may all contribute to
lay the groundwork for the development of myeloid diseases.[7,9-15]
The subsequent CT may later favor additional hit development, such as
the acquisition of genetic and/or cytogenetic abnormalities, the
selection of abnormal hematopoietic clones (e.g. with TP53 mutations
and/or unfavorable karyotype) and changes in the bone marrow
microenvironment (BMM), resulting in t-MN onset.[8,16-19]
In this mini-review we synthesize recent findings about the involvement of BMM in MN de novo (MDS
and AML) and therapy-related pathogenesis with a deeper focus on the
role of bone marrow mesenchymal stem cells (BM-MSC). For all other
players involved in the pathogenesis of t-MN (inherited predisposition,
exposure to genotoxic agents, clonal selection and abnormal bone marrow
microenvironment), we refer you to two recent reviews.[8,16]
Mesenchymal Stem Cell: an Intriguing Cell Within the Bone Marrow Cellular Metropolis
Human
BM can be considered a cellular metropolis, composed of highly
vascularized multicellular tissue containing self‐renewing HSCs, which
generate progeny that progressively differentiates into mature myeloid,
erythroid, and lymphoid cells. These HSCs in the BM are surrounded by a
plethora of cellular (endothelial cells, osteo-lineage cells,
adipocytes, MSC, fibroblasts, macrophages, neutrophils, megakaryocytes,
and immune cells) and noncellular (extracellular matrix and soluble
factors) components. BMM cells form distinctively organized niches
(endosteal, perivascular, arteriolar, and central medullary), with each
of these anatomical regions in the bone having a specialized role in
maintaining the quiescence, homing and mobilization of the HSC.[20-24]
Collectively, these different cell types interact with each other and
HSC both through direct contact‐based regulation and the secretion of
key signaling molecules and, in this way, participate in the
maintenance of hematopoietic homeostasis.[25]
In this
mini-review, we focus on mesenchymal stem cells, one of the players
involved in bone marrow homeostasis. Mesenchymal stem cells or
mesenchymal stromal cells are multipotent stem cells of mesodermal
origin (Figure 1) that can be isolated from adult and fetal tissues. In the bone marrow, they represent a rare population, accounting for 1/104 mononuclear cells. BM-MSCs have a fibroblast-like morphology. According to the International Society for Cellular Therapy,[26] a cell to be defined as a BM-MSC must comply with 3 minimum criteria:
- ability to adhere to a plastic substrate under standard culture conditions (unlike HSC grow in suspension)
-
immunophenotype positive for main mesenchymal markers such as CD73,
CD90, CD105 and negative for main hematopoietic markers such as CD14,
CD79, CD34, CD45, HLA-DR
- trilinear differentiative potential (osteogenic, adipogenicity and chondrogenic lineages).
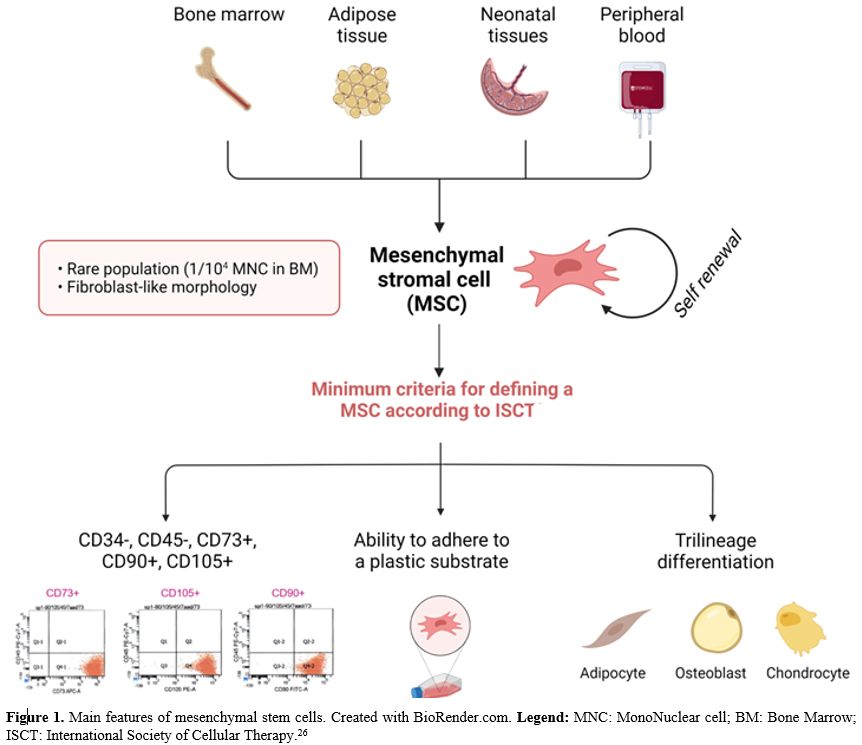 |
- Figure
1. Main features of mesenchymal stem cells. Created with BioRender.com.
Legend: MNC: MonoNuclear cell; BM: Bone Marrow; ISCT: International
- Society of Cellular Therapy.[26]
|
MSCs
are distinct from BM stromal cells, which are mostly comprised of
hematopoietic supporting fibroblasts, differentiated from MSC. In BM,
MSCs can be located at different anatomical sites (central sinus,
trabeculae, endosteal region, and compact bone). These locations are
also sites of hematopoietic activity in which the function of HSC is
supported by BM-MSC and their differentiated cells (e.g. fibroblasts,
adipocytes and osteoblasts). Thus, the functional relationship between
MSC and hematopoietic activity are part of the process of maintaining
BM homeostasis.
Physiologically, BM-MSCs have dual functions (Figure 2):
support hematopoiesis and regulate, by inhibiting, the immune response.
BM-MSCs regulate the balance between self-renewal and differentiation
of HSC through the production of various soluble factors (such as
growth factors and cytokines) as well as surface molecules.
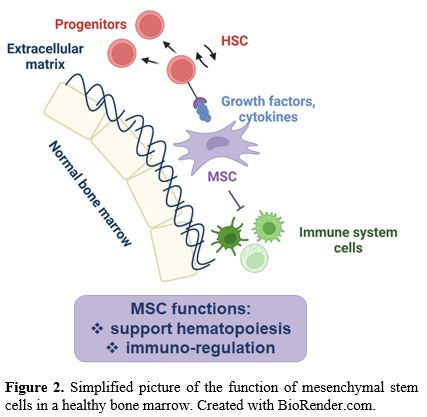 |
- Figure
2. Simplified picture of the function of mesenchymal stem cells in a healthy bone marrow. Created with BioRender.com.
|
Moreover,
BM-MSCs have immunoregulatory properties by maintaining the stability
of the BM immune microenvironment and reducing the damage caused to HSC
by stress stimuli.
Involvement of BM-MSC in de novo MN Development
Recent
studies have highlighted the role of a complex bidirectional crosstalk
between HSC and the BMM in normal hematopoiesis, as well as in the
pathogenesis of myeloid diseases.[27] Emerging data suggest that alterations of BM-MSC, an important component of the BMM,[28] may play a role in the pathogenesis of myeloid neoplasms, both de novo and therapy-related,[29-36] although the mechanisms are not yet fully understood.
The
first experimental evidence supporting the crucial role of BMM and MSC
in the initiation and progression of myeloid malignancies derived from
in vivo models. Using murine genetic models, several groups have shown
that specific genetic changes in the microenvironment, including
reduced function of genes such as RAR-γ, Rb, Mib1, IκBα, Sipa1, Dicer1
and concordant loss of the EGR1, APC, and TP53 in non-hematopoietic
cells, may have a pathophysiological significance in the genesis of
hematological malignancies, in particular for the creation of a
premetastatic niche that supports the growth and spread of clonal
neoplastic cells.[37-41]
In a mouse model of pre-leukemia, Zambetti and colleagues[42]
established a concept of mesenchymal niche-induced genotoxic stress in
HSC, providing conceptual and mechanistic insights into the link
between inflammation and cancer. The authors showed that perturbation
of mesenchymal cells in a mouse model of the pre-leukemic disorder
Shwachman-Diamond syndrome induces mitochondrial dysfunction, oxidative
stress, and activation of DNA damage responses (DDR) in HSPC through
p53-S100A8/9-TLR4 inflammatory signaling as a common driving mechanism
of genotoxic stress.[42]
Taken together, all
these mouse model studies strongly support the hypothesis that an
altered BMM provides ''fertile ground'' for the expansion of neoplastic
cells in vivo.
Moreover, BM-MSCs influence leukemic cells and are
essential for the propagation of human MDS-HSC in vivo in xenograft
models. Medyouf and colleagues showed the inability of human MDS stem
cells to propagate in a cell-autonomous manner and demonstrated that
co-injection of MDS-HSC with MDS-MSC in NSG mice results in a
significantly higher percentage of engraftment compared single
injection of MDS-HSC in the bone marrow of xenografted mice analyzed
16–28 weeks post-transplant.[33] Therefore, this
patient-derived xenograft model provides functional and molecular
evidence that MN is a complex disease that involves both the
hematopoietic and stromal compartments. An independent study has also
demonstrated that multiplex gene editing to confer leukemic drivers in
healthy human HSPC is insufficient for the development of leukemia
after transplantation in mice, supporting the need for a dysplastic
stroma in disease initiation.[43]
In summary,
BM-MSC may influence hematopoietic cells and similarly, hematopoietic
cells can induce remodeling of BM-MSC. After long periods of exposition
to neoplastic hematopoietic cells, healthy BM-MSCs can be reprogrammed,
acquiring functional alterations, to work in cooperation with leukemic
cells and propagate the disease.
Compared to BM-MSC isolated from
healthy donors (HD), BM-MSC isolated from patients with de novo MDS/AML
are structurally, epigenetically and functionally altered (Figure 3).[29-36]
Unlike the BM-MSC isolated from HD having the characteristic
fibroblast-like appearance, patient-derived BM-MSC present an altered
morphology, are larger and appear flattened and disorganized. Moreover,
they exhibit decreased proliferative and clonogenic capacity, reduced
osteogenic differentiation, increased senescence and impaired
immunoregulatory properties.[29-36]
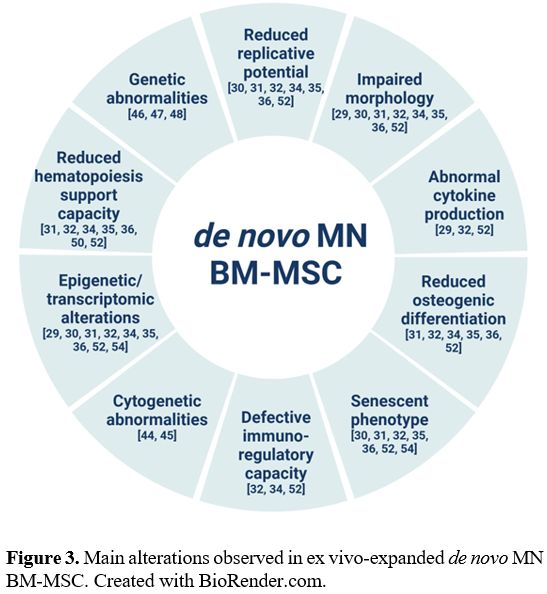 |
- Figure 3.. Main alterations observed in ex vivo-expanded de novo MN BM-MSC. Created with BioRender.com.
|
The
clonal origins of MN-MSC have always been questioned. There are
contradictory reports about the presence of ‘MN related-gene’ mutations
or chromosomal abnormalities in MN MSC.[44-51]
Regarding chromosomal analysis, Blau and colleagues reported BM-MSC
karyotype abnormalities in a fraction of MDS/AML patients (15-30%), but
not in healthy controls. Of note, these studies reported the occurrence
of non-clonal chromosomal aberrations in BM-MSC isolated from patients
with MDS and AML, which only very rarely correspond to the cytogenetic
markers observed in the hematopoietic leukemic clone of the same
individual.[44-45] In the same line, our group
investigated the frequency of recurrent mutations of epigenetic and
spliceosome genes in paired bone marrow hematopoietic and mesenchymal
cells isolated from patients with different myeloid malignancies. We
found no mutations for any of the studied genes in the MSC compartment,
both in carriers of mutations in the hematopoietic compartment and in
wild-type patients.[46]
Furthermore, MN–MSCs
have an altered expression of key molecules involved in the interaction
with HSPC, in particular Osteopontin, Jagged1, Kit-ligand and
Angiopoietin as well as several chemokines. [32,52]
Functionally, this translated into a significantly reduced ability of
MN-MSCs to support normal HSC in long-term culture-initiating cell
assays. When MDS HSC are cultured with MSC from healthy donors, their
clonogenic capacity is partially restored.[32] These
data indicate that diseased bone marrow cells are likely to play an
active role in the ‘‘reprogramming’’ of their BM niche during disease
development and/or progression by possibly converting it into a
self-supportive one.
A similar reduction in hematopoiesis support
can be reproduced by silencing the transcription factor FOXM1 in
BM-MSC. The repression of FOXM1, the transcription factor that drives
G2/M transition, in elderly mitotic cells, increased chromosome
mis-segregation and correlates with an early senescence-associated
phenotype.[53] Recently, our group have showed that
the silencing of FOXM1 mRNA in HD-MSCs recapitulates the downregulation
of FOXM1 and its mitotic targets observed in MSC de novo and
therapy-related MDS.[54] Furthermore, FOXM1 silencing
is able to reduce the supportive capacity of hemopoiesis as
demonstrated by the reduction of granulocyte colonies numbers after
coculture of healthy HSC with MSCs in which FOXM1 was silenced.[54]
Involvement of BM-MSC in Therapy-related MN Development
For
just over a decade, we have known that t-MN development is a
multifactorial process resulting from complex interactions between an
underlying germline genetic susceptibility, the stepwise acquisition of
somatic mutations in HSC, the clonal selection pressure exerted by CT
and alterations in the BMM.[8] However, although in the literature many works comparing the mutational/transcriptomic/epigenetic/cytogenetic profiling of de novo and therapy-related MN at the level of the hematopoietic compartment (HSC or HSPC) are present,[55-59] extensive studies concerning the involvement of BM-MSC in the pathogenesis of t-MN are only recently emerging.[30,46,60]
One of the first works in this regard was conducted by our group in
2016 on a cohort of patients with multiple hematologic malignancies,
including de novo MDS/AML and therapy-related myeloid neoplasms of whom we isolated and ex vivo-expanded BM-MSC.[30] In t-MN BM-MSCs, we observed an altered morphology and a decreased proliferative and clonogenic potential compared to HD-MSC.[30] Moreover, no mutations in genes involved in splicing, DNA methylation, and the TP53 gene have been identified in t-MN MSC.[46,60]
More
recently, to better decipher the microenvironmental changes induced by
CT vs. neoplasia, Kutyna and colleagues performed a multi-omic
(transcriptome, DNA damage response, cytokine secretome, and functional
profiling) characterization of BM-MSC both from patients with t-MN, MN,
and another cancer but without cytotoxic exposure, typical primary MN,
and age-matched controls.[60] The authors showed that
t-MN MSCs are distinct from HD-MSCs but are also distinct from other
primary MNs, developing apart from cytotoxic exposure. Strikingly,
among all studied populations, t-MN appeared to have the greatest
defect in terms of morphology, proliferative capacity, and support to
hematopoiesis.
What is the role of cytotoxic therapy in this
context? The role of CT is complex and not yet clear. Cytotoxic therapy
has been shown to exert several effects on the BMM, including a
pro-inflammatory response with the consequent release of inflammatory
cytokines (e.g., TNFα, TGFβ, and IL-6) and release of reactive oxygen
species (ROS) by MSC with resultant genotoxic damage to HSC.[8,41]
Stoddart
and colleagues described cooperative effects of exposing both the BMM
of recipient mice and donor HSPC to the alkylating agent
N-ethyl-N-nitrosourea (ENU) in a genetically model of therapy-induced
MDS and AML characterized by chromosome 5q deletions.[41]
In detail, the haploinsufficiency of two del(5q) genes (EGR1 and APC),
together with TP53 knockdown, in a mouse model, produces a high
frequency of myeloid diseases following concurrent treatment of both
hematopoietic cells and the BM stroma with ENU, but not after treatment
of either alone.[41] In addition, loss of TP53 with
EGR1 and APC was required to drive the development of a transplantable
leukemia and accompanied by the acquisition of somatic mutations in DDR
genes. ENU treatment of MSC induced cellular senescence and led to the
acquisition of a senescence-associated secretory phenotype (SASP),
which is a critical microenvironmental alteration in the pathogenesis
of t-MN.[41,56]
Similarly,
t-MN ex vivo expanded BM-MSC showed a profoundly senescent phenotype
with a characteristic flattened morphology, defective regenerative
capacity, high p21 and β-Galactosidase expression, and a SASP with
secretion of pro-inflammatory cytokines, chemokines, and proteases.[60]
Interestingly, the level of senescence in t-MN BM-MSCs was independent
of the latency period, the interval between completion of CT and t-MN
diagnosis. High levels of senescence were evident both in t-MN BM-MSC
with short (3–4 months) and long latency (up to two decades following
CT).[60] Moreover, BM-MSCs derived from t-MN had
higher baseline DNA damage and higher intracellular ROS levels compared
to HD BM-MSC and were highly sensitive to CT (e.g., Doxorubicin).[60]
Recently,
Özdemir and colleagues showed that alterations in the BM niche may play
a critical/driver role in the development of secondary AML. The
treatment with the chemotherapeutic agent Etoposide of HD BM-MSC is
able to induce an increased expression of selected genes involved in
xenobiotic metabolism, DNA double-strand break response, heat shock
response, and cell cycle regulation such as CYP1A1, GAD34, ATF4, NUPR1,
CXCL12, KLF4, CCNB1.[61]
Similarly, the high
senescence level observed in t-MN BM-MSCs is due to a defect in the DDR
pathway, resulting in permanent DNA damage after exposure to cytotoxic
therapy.[60,62] Sequential patient
sampling showed that exposure to DNA-damaging agents leads to
pro-inflammatory stromal defects and irreversible damage evident many
years before the onset and diagnosis of t-MN. These data underscore the
role of senescence in the pathogenesis of t-MN and provide a valuable
resource for future therapeutics with repercussions for patients
treated with chemotherapy or radiotherapy.
Despite their dormant
state, t-MN stromal cells were metabolically highly active with a
switch toward glycolysis and secreted multiple pro-inflammatory
cytokines (IFNγ, IL-7, IL-1β, IL-13, IL-15, and EGF) indicative of a
senescent-secretory phenotype that inhibited adipogenesis.[60]
t-MN MSC exhibited a selective defect in adipocyte differentiation that
was experimentally mimed by treating healthy BM-MSC with
senescence-secreted cytokines IL-1β and IFNγ. Treatment of HD BM-MSC
with IL-1β, IFNγ, IFNα, or a cocktail of cytokines (IL-1β, IL-13,
IL-15, IL-6, IFNα, and IFNγ) profoundly inhibited adipogenesis in
vitro, demonstrating a potential causative role of senescence-secreted
cytokines in inhibiting adipogenesis. These data suggest that the
secretome is modifying stromal fate.[60]
Finally,
Kutyna and colleagues showed that senolytic agents Dasatinib and
Quercetin alone or in combination effectively reduced the senescence
burden and restored the differentiation potential of t-MN BM-MSC,
indicating a possible role of senolytic therapies in modulating t-MN
long-term. Senolytics, including Dasatinib and Quercetin, have been
shown to selectively eliminate senescent cells from both humans and
mouse,[63-67] with evidence that sufficient restoration of function may occur without eliminating all senescent cells.[63,65,68] Indeed, in the study of Kutyna and colleagues, senolytics restored the defect in adipogenesis differentiation in t-MN.
Currently,
there is an enhanced focus on extrinsic, age-related changes in the BM
microenvironment that accompany the development of t-MN. One of the
most prominent changes associated with aging is the accumulation of
senescent BM-MSC within tissues and organs. In comparison with
proliferating cells, senescent cells display an altered secretome
comprising proteases, inflammatory cytokines, and growth factors that
may render the local microenvironment favorable for cancer growth.[69]
There is emerging evidence that BM-MSC senescence may contribute to
age-related hematopoietic decline and cancer development. Moreover, CT
creates an environment that selects for pre-existing mutant clones at
the expense of normal HSCs. In this context, DNA damage-induced
competition led to a selective clonal advantage of HSCs and
hematopoietic progenitor cells with reduced p53 function in mouse BM
chimeras, reminiscent of the CHIP phenotype, via growth arrest and
senescence-related gene expression in cells with higher p53 activity.[70]
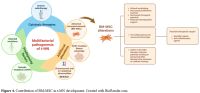 |
- Figure 4.. Contribution of BM-MSC in t-MN development. Created with BioRender.com.
|
Summary and Future Prospective
Therapy-related
myeloid neoplasms are a multifactorial disease resulting from complex
interactions between a germline genetic susceptibility, the acquisition
of somatic mutations in hematopoietic stem cells, the clonal pressure
exerted by cytotoxic therapies, and alterations of the bone marrow
microenvironment.
BM-MSC isolated from patients with t-MN present
several alterations (e.g., pro-inflammatory and senescent phenotype),
both intrinsic and extrinsic, contributing to the pathogenesis of t-MN
and could provide a valuable resource for future therapeutics. It would
be interesting to understand whether the highly pro-inflammatory SASP
observed in BM-MSC-derived from t-MN could initiate or promote a form
of clonal hematopoiesis, eventually progressing to t-MN and whether it
could become an effective therapeutic target.
References
- Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R,
Apperley JF, Bejar R, Berti E, Busque L, Chan JKC, Chen W, Chen X, Chng
WJ, Choi JK, Colmenero I, Coupland SE, Cross NCP, De Jong D, Elghetany
MT, Takahashi E, Emile JF, Ferry J, Fogelstrand L, Fontenay M, Germing
U, Gujral S, Haferlach T, Harrison C, Hodge JC, Hu S, Jansen JH,
Kanagal-Shamanna R, Kantarjian HM, Kratz CP, Li XQ, Lim MS, Loeb K,
Loghavi S, Marcogliese A, Meshinchi S, Michaels P, Naresh KN, Natkunam
Y, Nejati R, Ott G, Padron E, Patel KP, Patkar N, Picarsic J,
Platzbecker U, Roberts I, Schuh A, Sewell W, Siebert R, Tembhare P,
Tyner J, Verstovsek S, Wang W, Wood B, Xiao W, Yeung C, Hochhaus A. The
5th edition of the World Health Organization Classification of
Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms.
Leukemia. 2022;36:1703-19. https://doi.org/10.1038/s41375-022-01613-1 PMid:35732831 PMCid:PMC9252913
- Arber
DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM,
Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World
Health Organization classification of myeloid neoplasms and acute
leukemia. Blood. 2016;127:2391-405. https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Vardiman
JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL,
Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008
revision of the World Health Organization (WHO) classification of
myeloid neoplasms and acute leukemia: rationale and important changes.
Blood. 2009;114:937-51. https://doi.org/10.1182/blood-2009-03-209262 PMid:19357394
- Guru
Murthy GS, Hamadani M, Dhakal B, Hari P, Atallah E. Incidence and
survival of therapy related myeloid neoplasm in United States. Leuk
Res. 2018;71:95-99. https://doi.org/10.1016/j.leukres.2018.07.013 PMid:30048839
- Gurnari
C, Fabiani E, Falconi G, Travaglini S, Ottone T, Cristiano A, Voso MT.
From Clonal Hematopoiesis to Therapy-Related Myeloid Neoplasms: The
Silent Way of Cancer Progression. Biology (Basel). 2021;10:128. https://doi.org/10.3390/biology10020128 PMid:33562056 PMCid:PMC7914896
- Fianchi
L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M, Criscuolo M,
Specchia G, Maria Pogliani E, Maurillo L, Aloe-Spiriti MA, Mecucci C,
Niscola P, Rossetti E, Mansueto G, Rondoni M, Fozza C, Invernizzi R,
Spadea A, Fenu S, Buda G, Gobbi M, Fabiani E, Sica S, Hohaus S, Leone
G, Voso MT. Characteristics and outcome of therapy-related myeloid
neoplasms: Report from the Italian network on secondary leukemias. Am J
Hematol. 2015;90:E80-5. Epub 2015 Mar 3. https://doi.org/10.1002/ajh.23966 PMid:25653205
- Desai P, Roboz GJ. Clonal Hematopoiesis and therapy related MDS/AML. Best Pract Res Clin Haematol. 2019;32:13-23. https://doi.org/10.1016/j.beha.2019.02.006 PMid:30927970
- McNerney
ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when
genetics and environment collide. Nat Rev Cancer. 2017;17:513-27. https://doi.org/10.1038/nrc.2017.60 PMid:28835720 PMCid:PMC5946699
- Swaminathan
M, Bannon SA, Routbort M, Naqvi K, Kadia TM, Takahashi K, Alvarado Y,
Ravandi-Kashani F, Patel KP, Champlin R, Kantarjian H, Strong L,
DiNardo CD. Hematologic malignancies and Li-Fraumeni syndrome. Cold
Spring Harb Mol Case Stud. 2019;5:a003210. Print 2019 Feb. https://doi.org/10.1101/mcs.a003210 PMid:30709875 PMCid:PMC6371746
- Schwartz
JR, Ma J, Kamens J, Westover T, Walsh MP, Brady SW, Robert Michael J,
Chen X, Montefiori L, Song G, Wu G, Wu H, Branstetter C, Hiltenbrand R,
Walsh MF, Nichols KE, Maciaszek JL, Liu Y, Kumar P, Easton J, Newman S,
Rubnitz JE, Mullighan CG, Pounds S, Zhang J, Gruber T, Ma X, Klco JM.
The acquisition of molecular drivers in pediatric therapy-related
myeloid neoplasms. Nat Commun. 2021;12:985. https://doi.org/10.1038/s41467-021-21255-8 PMid:33579957 PMCid:PMC7880998
- Berger
G, van den Berg E, Sikkema-Raddatz B, Abbott KM, Sinke RJ, Bungener LB,
Mulder AB, Vellenga E. Re-emergence of acute myeloid leukemia in donor
cells following allogeneic transplantation in a family with a germline
DDX41 mutation. Leukemia. 2017;31:520-2. https://doi.org/10.1038/leu.2016.310 PMid:27795557
- Baliakas
P, Tesi B, Wartiovaara-Kautto U, Stray-Pedersen A, Friis LS, Dybedal I,
Hovland R, Jahnukainen K, Raaschou-Jensen K, Ljungman P, Rustad CF,
Lautrup CK, Kilpivaara O, Kittang AO, Grønbæk K, Cammenga J,
Hellström-Lindberg E, Andersen MK. Nordic Guidelines for Germline
Predisposition to Myeloid Neoplasms in Adults: Recommendations for
Genetic Diagnosis, Clinical Management and Follow-up. Hemasphere.
2019;3:e321. eCollection 2019 Dec. https://doi.org/10.1097/HS9.0000000000000321 PMid:31976490 PMCid:PMC6924562
- Jaiswal
S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley
RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC,
Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G,
Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy
MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG,
Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis
associated with adverse outcomes. N Engl J Med. 2014;371:2488-98. https://doi.org/10.1056/NEJMoa1408617 PMid:25426837 PMCid:PMC4306669
- Genovese
G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert
K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M,
Höglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF,
Sklar P, Grönberg H, Hultman CM, McCarroll SA. Clonal hematopoiesis and
blood-cancer risk inferred from blood DNA sequence. N Engl J Med.
2014;371:2477-87. https://doi.org/10.1056/NEJMoa1409405 PMid:25426838 PMCid:PMC4290021
- Wong
TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL,
Shen D, Hundal J, Fulton RS, Heath S, Baty JD, Klco JM, Ding L, Mardis
ER, Westervelt P, DiPersio JF, Walter MJ, Graubert TA, Ley TJ, Druley
T, Link DC, Wilson RK. Role of TP53 mutations in the origin and
evolution of therapy-related acute myeloid leukaemia. Nature.
2015;518:552-5. https://doi.org/10.1038/nature13968 PMid:25487151 PMCid:PMC4403236
- Voso
MT, Falconi G, Fabiani E. What's new in the pathogenesis and treatment
of therapy-related myeloid neoplasms. Blood. 2021;138:749-57. https://doi.org/10.1182/blood.2021010764 PMid:33876223
- Fabiani
E, Falconi G, Fianchi L, Criscuolo M, Ottone T, Cicconi L, Hohaus S,
Sica S, Postorino M, Neri A, Lionetti M, Leone G, Lo-Coco F, Voso MT.
Clonal evolution in therapy-related neoplasms. Oncotarget.
2017;8:12031-40. https://doi.org/10.18632/oncotarget.14509 PMid:28076841 PMCid:PMC5355323
- Voso
MT, Pandzic T, Falconi G, Denčić-Fekete M, De Bellis E, Scarfo L,
Ljungström V, Iskas M, Del Poeta G, Ranghetti P, Laidou S, Cristiano A,
Plevova K, Imbergamo S, Engvall M, Zucchetto A, Salvetti C, Mauro FR,
Stavroyianni N, Cavelier L, Ghia P, Stamatopoulos K, Fabiani E,
Baliakas P. Clonal haematopoiesis as a risk factor for therapy-related
myeloid neoplasms in patients with chronic lymphocytic leukaemia
treated with chemo-(immuno)therapy. Br J Haematol. 2022;198:103-13. https://doi.org/10.1111/bjh.18129 PMid:35277855
- Renneville
A, Bernard E, Micol JB. Therapy-related myelodysplastic syndromes in
the genomics era. Bull Cancer. 2023;28:S0007-4551(23)00277-1. https://doi.org/10.1016/j.bulcan.2023.02.022 PMid:37391357
- Batsivari
A, Grey W, Bonnet D. Understanding of the crosstalk between normal
residual hematopoietic stem cells and the leukemic niche in acute
myeloid leukemia. Exp. Hematol. 2021;95:23-30. https://doi.org/10.1016/j.exphem.2021.01.004 PMid:33497761
- Scadden DT. Nice neighborhood: Emerging concepts of the stem cell niche. Cell. 2014; 157:41-50. https://doi.org/10.1016/j.cell.2014.02.013 PMid:24679525 PMCid:PMC4161226
- Pinho
S, Frenette PS. Haematopoietic stem cell activity and interactions with
the niche. Nat. Rev. Mol. Cell Biol. 2019;20:303-20. https://doi.org/10.1038/s41580-019-0103-9 PMid:30745579 PMCid:PMC6483843
- Goulard M, Dosquet C, Bonnet D. Role of the microenvironment in myeloid malignancies. Cell Mol. Life Sci. 2018;75:1377-91 https://doi.org/10.1007/s00018-017-2725-4 PMid:29222645 PMCid:PMC5852194
- Mian
SA, Bonnet D. Nature or Nurture? Role of the Bone Marrow
Microenvironment in the Genesis and Maintenance of Myelodysplastic
Syndromes. Cancers (Basel). 2021;13:4116. https://doi.org/10.3390/cancers13164116 PMid:34439269 PMCid:PMC8394536
- Ghobrial
IM, Detappe A, Anderson KC, Steensma DP. The bone-marrow niche in MDS
and MGUS: implications for AML and MM. Nat Rev Clin Oncol.
2018;15:219-33. https://doi.org/10.1038/nrclinonc.2017.197 PMid:29311715
- Dominici
M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D,
Deans R, Keating A, Prockop Dj, Horwitz E. Minimal criteria for
defining multipotent mesenchymal stromal cells. The International
Society for Cellular Therapy position statement. Cytotherapy.
2006;8:315-7. https://doi.org/10.1080/14653240600855905 PMid:16923606
- Agarwal
P, Bhatia R. Influence of bone marrow microenvironment on leukemic stem
cells: breaking up an intimate relationship. Adv Cancer Res.
2015;127:227-52. https://doi.org/10.1016/bs.acr.2015.04.007 PMid:26093902
- Bulycheva
E, Rauner M, Medyouf H, Theurl I, Bornhäuser M, Hofbauer LC,
Platzbecker U. Myelodysplasia is in the niche: novel concepts and
emerging therapies. Leukemia. 2015;29:259-68. https://doi.org/10.1038/leu.2014.325 PMid:25394715 PMCid:PMC4320287
- Aanei
CM, Flandrin P, Eloae FZ, Carasevici E, Guyotat D, Wattel E, Campos L.
Intrinsic growth deficiencies of mesenchymal stromal cells in
myelodysplastic syndromes. Stem Cells Dev. 2012;21:1604-15. https://doi.org/10.1089/scd.2011.0390 PMid:21933023 PMCid:PMC3376465
- Falconi
G, Fabiani E, Fianchi L, Criscuolo M, Raffaelli CS, Bellesi S, Hohaus
S, Voso MT, D'Alò F, Leone G. Impairment of PI3K/AKT and WNT/β-catenin
pathways in bone marrow mesenchymal stem cells isolated from patients
with myelodysplastic syndromes. Exp Hematol. 2016;44:75-83. https://doi.org/10.1016/j.exphem.2015.10.005 PMid:26521017
- Fei
C, Zhao Y, Guo J, Gu S, Li X, Chang C. Senescence of bone marrow
mesenchymal stromal cells is accompanied by activation of p53/p21
pathway in myelodysplastic syndromes. Eur J Haematol. 2014;93:476-86. https://doi.org/10.1111/ejh.12385 PMid:24889123
- Geyh
S, Oz S, Cadeddu RP, Fröbel J, Brückner B, Kündgenet A, Fenk R, Bruns
I, Zilkens C, Hermsen D, Gattermann N, Kobbe G, Germing U, Lyko F, Haas
R, Schroeder T. Insufficient stromal support in MDS results from
molecular and functional deficits of mesenchymal stromal cells.
Leukemia. 2013;27:1841-51. https://doi.org/10.1038/leu.2013.193 PMid:23797473
- Medyouf
H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, Lier A, Eisen C,
Nowak V, Zens B, Müdder K, Klein C, Obländer J, Fey S, Vogler J,
Fabarius A, Riedl E, Roehl H, Kohlmann A, Staller M, Haferlach C,
Müller N, John T, Platzbecker U, Metzgeroth G, Hofmann WK, Trumpp A,
Nowak D. Myelodysplastic cells in patients reprogram mesenchymal
stromal cells to establish a transplantable stem cell niche disease
unit. Cell Stem Cell. 2014;14:824-37. https://doi.org/10.1016/j.stem.2014.02.014 PMid:24704494
- Zhao
ZG, Xu W, Yu HP, Fang BL, Wu SH, Li F, Li WM, Li QB, Chen ZC, Zou P.
Functional characteristics of mesenchymal stem cells derived from bone
marrow of patients with myelodysplastic syndromes. Cancer Lett.
2012;317:136-43. https://doi.org/10.1016/j.canlet.2011.08.030 PMid:22240014
- Desbourdes
L, Javary J, Charbonnier T, Ishac N, Bourgeais J, Iltis A, Chomel JC,
Turhan A, Guilloton F, Tarte K, Demattei MV, Ducrocq E, Rouleux-Bonnin
F, Gyan E, Hérault O, Domenech J. Alteration Analysis of Bone Marrow
Mesenchymal Stromal Cells from De Novo Acute Myeloid Leukemia Patients
at Diagnosis. Stem Cells Dev. 2017;26:709-22. https://doi.org/10.1089/scd.2016.0295 PMid:28394200
- von
der Heide EK, Neumann M, Vosberg S, James AR, Schroeder MP,
Ortiz-Tanchez J, Isaakidis K, Schlee C, Luther M, Jöhrens K,
Anagnostopoulos I, Mochmann LH, Nowak D, Hofmann WK, Greif PA, Baldus
CD. Molecular alterations in bone marrow mesenchymal stromal cells
derived from acute myeloid leukemia patients. Leukemia.
2017;31:1069-78. https://doi.org/10.1038/leu.2016.324 PMid:27833093
- Raaijmakers
MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL,
Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M,
Merkenschlager M, Lin C, Rommens JM, Scadden DT. Bone progenitor
dysfunction induces myelodysplasia and secondary leukaemia. Nature.
2010;464:852-7. https://doi.org/10.1038/nature08851 PMid:20305640 PMCid:PMC3422863
- Walkley
CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland
SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced
myeloproliferative syndrome caused by retinoic acid receptor gamma
deficiency. Cell 2007;129:1097-110. https://doi.org/10.1016/j.cell.2007.05.014 PMid:17574023 PMCid:PMC1974882
- Rupec
RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, Flaig MJ,
Moosmann S, Plewig G, Dörken B, Förster I, Huss R, Pfeffer K. Stroma
mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha.
Immunity 2005;22:479-91. https://doi.org/10.1016/j.immuni.2005.02.009 PMid:15845452
- Xiao
P, Dolinska M, Sandhow L, Kondo M, Johansson AS, Bouderlique T, Zhao Y,
Li X, Dimitriou M, Rassidakis GZ, Hellström-Lindberg E, Minato N,
Walfridsson J, Scadden DT, Sigvardsson M, Qian H. Sipa1
deficiency-induced bone marrow niche alterations lead to the initiation
of myeloproliferative neoplasm. Blood Adv. 2018;2:534-48. https://doi.org/10.1182/bloodadvances.2017013599 PMid:29514790 PMCid:PMC5851419
- Stoddart
A, Wang J, Fernald AA, Davis EM, Johnson CR, Hu C, Cheng JX, McNerney
ME, Le Beau MM. Cytotoxic Therapy-Induced Effects on Both Hematopoietic
and Marrow Stromal Cells Promotes Therapy-Related Myeloid Neoplasms.
Blood Cancer Discovery. 2020;1:32-47. https://doi.org/10.1158/2643-3230.BCD-19-0028 PMid:32924016 PMCid:PMC7486063
- Zambetti
NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, Hoogenboezem
RM, Bindels EMJ, Adisty MN, Van Strien PMH, van der Leije CS, Westers
TM, Cremers EMP, Milanese C, Mastroberardino PG, van Leeuwen JPTM, van
der Eerden BCJ, Touw IP, Kuijpers TW, Kanaar R, van de Loosdrecht AA,
Vogl T, Raaijmakers MHGP. Mesenchymal Inflammation Drives Genotoxic
Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in
Human Pre-leukemia. Cell Stem Cell. 2016;19:613-27. https://doi.org/10.1016/j.stem.2016.08.021 PMid:27666011
- Tothova
Z, Krill-Burger JM, Popova KD, Landers CC, Sievers QL, Yudovich D,
Belizaire R, Aster JC, Morgan EA, Tsherniak A, Ebert BL. Multiplex
CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells
Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell.
2017;21:547-555.e8. https://doi.org/10.1016/j.stem.2017.07.015 PMid:28985529 PMCid:PMC5679060
- Blau
O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schümann E, Thiel E, Blau
IW. Chromosomal aberrations in bone marrow mesenchymal stroma cells
from patients with myelodysplastic syndrome and acute myeloblastic
leukemia. Exp Hematol. 2007;35:221-9. https://doi.org/10.1016/j.exphem.2006.10.012 PMid:17258071
- Blau
O, Baldus CD, Hofmann WK, Thiel G, Nolte F, Burmeister T, Türkmen S,
Benlasfer O, Schümann E, Sindram A, Molkentin M, Mundlos S, Keilholz U,
Thiel E, Blau IW. Mesenchymal stromal cells of myelodysplastic syndrome
and acute myeloid leukemia patients have distinct genetic abnormalities
compared with leukemic blasts. Blood. 2011;118:5583-92. https://doi.org/10.1182/blood-2011-03-343467 PMid:21948175 PMCid:PMC3217359
- Fabiani
E, Falconi G, Fianchi L, Guidi F, Bellesi S, Voso MT, Leone G, D'Alò F.
Mutational analysis of bone marrow mesenchymal stromal cells in myeloid
malignancies. Exp Hematol. 2014;42:731-3. https://doi.org/10.1016/j.exphem.2014.04.011 PMid:24796317
- Jann
JC, Mossner M, Riabov V, Altrock E, Schmitt N, Flach J, Xu Q, Nowak V,
Obländer J, Palme I, Weimer N, Streuer A, Jawhar A, Darwich A, Jawhar
M, Metzgeroth G, Nolte F, Hofmann WK, Nowak D. Bone marrow derived
stromal cells from myelodysplastic syndromes are altered but not
clonally mutated in vivo. Nat Commun. 2021;12:6170. https://doi.org/10.1038/s41467-021-26424-3 PMid:34697318 PMCid:PMC8546146
- Azuma
K, Umezu T, Imanishi S, Asano M, Yoshizawa S, Katagiri S, Ohyashiki K,
Ohyashiki JH. Genetic variations of bone marrow mesenchymal stromal
cells derived from acute leukemia and myelodysplastic syndrome by
targeted deep sequencing. Leuk Res. 2017;62:23-28. https://doi.org/10.1016/j.leukres.2017.09.008 PMid:28964959
- Poon
Z, Dighe N, Venkatesan SS, Cheung AMS, Fan X, Bari S, Hota M, Ghosh S,
Hwang WYK. Bone marrow MSCs in MDS: contribution towards dysfunctional
hematopoiesis and potential targets for disease response to
hypomethylating therapy. Leukemia. 2019;33:1487-1500. https://doi.org/10.1038/s41375-018-0310-y PMid:30575819 PMCid:PMC6756222
- Corradi
G, Baldazzi C, Očadlíková D, Marconi G, Parisi S, Testoni N, Finelli C,
Cavo M, Curti A, Ciciarello M. Mesenchymal stromal cells from
myelodysplastic and acute myeloid leukemia patients display in vitro
reduced proliferative potential and similar capacity to support
leukemia cell survival. Stem Cell Res Ther. 2018;9:271. https://doi.org/10.1186/s13287-018-1013-z PMid:30359303 PMCid:PMC6202844
- Choi
H, Kim Y, Kang D, Kwon A, Kim J, Min Kim J, Park SS, Kim YJ, Min CK,
Kim M. Common and different alterations of bone marrow mesenchymal
stromal cells in myelodysplastic syndrome and multiple myeloma. Cell
Prolif. 2020;53:e12819. Epub 2020 May 5. https://doi.org/10.1111/cpr.12819
- Geyh
S, Rodríguez-Paredes M, Jäger P, Khandanpour C, Cadeddu RP, Gutekunst
J, Wilk CM, Fenk R, Zilkens C, Hermsen D, Germing U, Kobbe G, Lyko F,
Haas R, Schroeder T. Functional inhibition of mesenchymal stromal cells
in acute myeloid leukemia. Leukemia. 2016;30:683-91. https://doi.org/10.1038/leu.2015.325 PMid:26601782
- Macedo
JC, Vaz S, Bakker B, Ribeiro R, Bakker PL, Escandell JM, Ferreira MG,
Medema R, Foijer F, Logarinho E. FoxM1 repression during human aging
leads to mitotic decline and aneuploidy-driven full senescence. Nat
Commun. 2018;9:2834. https://doi.org/10.1038/s41467-018-05258-6 PMid:30026603 PMCid:PMC6053425
- Falconi,
G., Galossi, E., Fabiani, E., Pieraccioli, M., Travaglini, S.,
Hajrullaj, H., Cerretti, R., Palmieri, R., Latagliata, R., Maurillo,
L., Voso, MT., 2022. Impairment of FOXM1 expression in mesenchymal
cells from patients with myeloid neoplasms, de novo and
therapy-related, may compromise their ability to support hematopoiesis.
Sci Rep 12(1):21231. https://doi.org/10.1038/s41598-022-24644-1 PMid:36481766 PMCid:PMC9732345
- Shannon K, Link DC. Soil and Seed: Coconspirators in Therapy-Induced Myeloid Neoplasms. Blood Cancer Discov. 2020;1:10-12. https://doi.org/10.1158/2643-3249.BCD-20-0080 PMid:34661135 PMCid:PMC8500704
- Leone
G, Fabiani E, Voso MT. De Novo and Therapy-Related Myelodysplastic
Syndromes: Analogies and Differences. Mediterr J Hematol Infect Dis.
2022;14:e2022030. https://doi.org/10.4084/MJHID.2022.030 PMid:35615324 PMCid:PMC9083943
- Ok
CY, Patel KP, Garcia-Manero G, Routbort MJ, Peng J, Tang G, Goswami M,
Young KH, Singh R, Medeiros LJ, Kantarjian HM, Luthra R, Wang SA. TP53
mutation characteristics in therapy-related myelodysplastic syndromes
and acute myeloid leukemia is similar to de novo diseases. J Hematol
Oncol. 2015;8:45. https://doi.org/10.1186/s13045-015-0139-z PMid:25952993 PMCid:PMC4431603
- Ok
CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, Goswami M,
Singh R, Kanagal-Shamanna R, Pierce SA, Young KH, Kantarjian HM,
Medeiros LJ, Luthra R, Wang SA. Mutational profiling of therapy-related
myelodysplastic syndromes and acute myeloid leukemia by next generation
sequencing, a comparison with de novo diseases. Leuk Res.
2015;39:348-54. https://doi.org/10.1016/j.leukres.2014.12.006 PMid:25573287 PMCid:PMC5548131
- Kanagal-Shamanna
R, Yin CC, Miranda RN, Bueso-Ramos CE, Wang XI, Muddasani R, Medeiros
LJ, Lu G. Therapy-related myeloid neoplasms with isolated del(20q):
comparison with cases of de novo myelodysplastic syndrome with
del(20q). Cancer Genet. 2013;206:42-6. https://doi.org/10.1016/j.cancergen.2012.12.005 PMid:23357231
- Kutyna
MM, Kok CH, Lim Y, Tran ENH, Campbell D, Paton S, Thompson-Peach C, Lim
K, Cakouros D, Arthur A, Hughes T, Kumar S, Thomas D, Gronthos S,
Hiwase DK. A senescence stress secretome is a hallmark of
therapy-related myeloid neoplasm stromal tissue occurring soon after
cytotoxic exposure. Leukemia. 2022;36:2678-89. https://doi.org/10.1038/s41375-022-01686-y PMid:36038666 PMCid:PMC9613466
- Özdemir
C, Muratoğlu B, Özel BN, Alpdündar-Bulut E, Tonyalı G, Ünal Ş,
Uçkan-Çetinkaya D. Multiparametric analysis of etoposide exposed
mesenchymal stem cells and Fanconi anemia cells: implications in
development of secondary myeloid malignancy. Clin Exp Med. 2023 May 13.
Online ahead of print. https://doi.org/10.1007/s10238-023-01087-0 PMid:37179284
- Gynn
LE, Anderson E, Robinson G, Wexler SA, Upstill-Goddard G, Cox C, May
JE. Primary mesenchymal stromal cells in co-culture with leukaemic
HL-60 cells are sensitised to cytarabine-induced genotoxicity, while
leukaemic cells are protected. Mutagenesis. 2021;36:419-28. https://doi.org/10.1093/mutage/geab033 PMid:34505878 PMCid:PMC8633936
- Zhu
Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer
AK, Ikeno Y, Hubbard GB, Lenburg M, O'Hara SP, LaRusso NF, Miller JD,
Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout
MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D,
Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD,
Niedernhofer LJ, Kirkland JL. The Achilles' heel of senescent cells:
from transcriptome to senolytic drugs. Aging Cell. 2015;14:644-58. https://doi.org/10.1111/acel.12344 PMid:25754370 PMCid:PMC4531078
- Suvakov
S, Cubro H, White WM, Butler Tobah YS, Weissgerber TL, Jordan KL, Zhu
XY, Woollard JR, Chebib FT, Milic NM, Grande JP, Xu M, Tchkonia T,
Kirkland JL, Lerman LO, Garovic VD. Targeting senescence improves
angiogenic potential of adipose-derived mesenchymal stem cells in
patients with preeclampsia. Biol Sex Differ. 2019;10:49. https://doi.org/10.1186/s13293-019-0263-5 PMid:31521202 PMCid:PMC6744626
- Farr
JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir
JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ,
Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S.
Targeting cellular senescence prevents age-related bone loss in mice.
Nat Med. 2017;23:1072-19. https://doi.org/10.1038/nm.4385 PMid:28825716 PMCid:PMC5657592
- Zhu
Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M,
Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T,
Kirkland JL. New agents that target senescent cells: the flavone,
fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging
2017;9:955-63. https://doi.org/10.18632/aging.101202 PMid:28273655 PMCid:PMC5391241
- Zoico
E, Nori N, Darra E, Tebon M, Rizzatti V, Policastro G, De Caro A, Rossi
AP, Fantin F, Zamboni M. Senolytic effects of quercetin in an in vitro
model of pre-adipocytes and adipocytes induced senescence. Sci Rep.
2021;11:23237. https://doi.org/10.1038/s41598-021-02544-0 PMid:34853352 PMCid:PMC8636588
- Lewis-McDougall
FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, Clark
JE, Punjabi PP, Awad W, Torella D, Tchkonia T, Kirkland JL,
Ellison-Hughes GM. Aged-senescent cells contribute to impaired heart
regeneration. Aging Cell. 2019;18:e12931. Epub 2019 Mar 10. https://doi.org/10.1111/acel.12931 PMid:30854802 PMCid:PMC6516154
- Plakhova
N, Panagopoulos V, Vandyke K, Zannettino ACW, Mrozik KM. Mesenchymal
stromal cell senescence in haematological malignancies. Cancer
Metastasis Rev. 2023;42:277-96. https://doi.org/10.1007/s10555-022-10069-9 PMid:36622509
- Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309-22. https://doi.org/10.1016/j.stem.2010.03.002 PMid:20362536 PMCid:PMC2872065