Viviane Lamim Lovatel1, Beatriz Ferreira da Silva1, Eliane Ferreira Rodrigues1, Maria Luiza Rocha da Rosa Borges2, Rita de Cássia Barbosa Tavares3, Ana Paula Silva Bueno4, Elaine Sobral da Costa4, Terezinha de Jesus Marques Salles2 and Teresa de Souza Fernandez1.
1 Cytogenetic Laboratory, Cell and Gene Therapy Program, Instituto Nacional do Câncer (INCA), Rio de Janeiro, RJ, Brazil.
2 Centro Oncohematologico Pediátrico, Hospital Universitário Oswaldo Cruz (HUOC), Recife, PE, Brazil.
3
Outpatient Department, Bone Marrow Transplantation Center (CEMO),
Instituto Nacional do Câncer (INCA), Rio de Janeiro, RJ, Brazil.
4
Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG),
Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ,
Brazil.
Correspondence to:
Teresa de Souza Fernandez. Praça da Cruz Vermelha 23, 6
o andar,
Laboratório de Citogenética, Centro de Transplante de Medula Óssea,
Instituto Nacional de Câncer (INCA), Rio de Janeiro, RJ, Brasil, CEP
20230-130. Phone: +55 (21)3207-1701. e-mail:
teresafernandez@inca.gov.br
Published: January 01, 2024
Received: September 04, 2023
Accepted: December 10, 2023
Mediterr J Hematol Infect Dis 2024, 16(1): e2024003 DOI
10.4084/MJHID.2024.003
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and objective:
Pediatric myelodysplastic syndrome (pMDS) is a group of rare clonal
neoplasms with a difficult diagnosis and risk of progression to acute
myeloid leukemia (AML). The early stratification in risk groups is
essential to choose the treatment and indication for allogeneic
hematopoietic stem cell transplantation (HSCT). According to the
Revised International Prognostic Scoring System, cytogenetic analysis
has demonstrated an essential role in diagnosis and prognosis. In pMDS,
abnormal karyotypes are present in 30-50% of the cases. Monosomy 7 is
the most common chromosomal alteration associated with poor prognosis.
However, the rarity of specific cytogenetic alterations makes its
prognosis uncertain. Thus, this study aimed to describe uncommon
cytogenetic alterations in a cohort of 200 pMDS patients and their
association with evolution to AML. Methods:
The cytogenetic analysis was performed in 200 pMDS patients by
G-banding and fluorescence in situ hybridization between 2000 to
2022. Results: Rare
chromosome alterations were observed in 7.5% (15/200) of the cases.
These chromosome alterations were divided into four cytogenetic groups:
hyperdiploidy, biclonal chromosomal alterations, translocations, and
uncommon deletions representing 33.3%, 33.3%, 20%, and 13.3%,
respectively. Most of these patients (10/15) were classified with
advanced MDS (MDS-EB and MDS/AML) and the initial subtype was present
in five patients (RCC). The leukemic evolution was observed in 66.66%
(10/15) of the patients. Most patients had poor clinical outcomes and
they were indicated for HSCT. Conclusion: The study of uncommon cytogenetic alterations in pMDS is important to improve the prognosis and guide early indication of HSCT.
|
Introduction
In recent years, significant advances in understanding myelodysplastic syndrome (MDS) pathology allowed a new classification.[1]
MDS is a heterogeneous group of clonal stem cell diseases with a risk
of evolution to acute myeloid leukemia (AML). Pediatric MDS (pMDS) is a
rare disease with a difficult diagnosis and variable clinical course,
and it is biologically distinct from adult MDS.[2] The
Revised International Prognostic Scoring System (IPSS-R) has been used
to stratify risk groups in pMDS. Thus, the cytogenetic analysis for
these patients is essential since the karyotypic pattern is one of the
pillars of IPSS-R.[3]
The incidence of
cytogenetic alterations in pMDS ranges from 30 to 50%, being more
frequent in advanced subtypes such as pMDS with excess blasts, MDS-EB
(5-19% of blasts), and MDS/AML (20-29% of blasts). The most frequent
cytogenetic alteration in pMDS is monosomy 7 (-7), which is associated
with a poor prognosis, followed by trisomy 8 (+8), and trisomy 21 (+21)
which have an intermediate prognosis and complex karyotypes with very
poor prognosis.[3,4] Rare chromosomal alterations such
as del(13q), -21, +11, +13, +14, +14q, and 11q23 translocations
were described in adult patients with MDS. These chromosomal
alterations do not have their real prognosis determined due to the low
number of cases described.[5,6] So, these uncommon
chromosomal alterations are classified mainly as an intermediate
cytogenetic risk group according to the IPSS-R.[3]
The
importance of refining the cytogenetic classification to determine the
prognostic significance of rare chromosomal abnormalities has been
pointed out in other diseases such as AML.[7] Since
the cytogenetic findings were the first diagnostic tool to stratify
patients and their prognosis, accurate stratification is essential to
choose the therapy and to indicate the hematopoietic stem cell
transplantation (HSCT), which is the only curative treatment for MDS
patients.[8,9] Nevertheless, to the best of our
knowledge, studies focusing on the frequency of uncommon chromosomal
alterations have not been previously performed in pMDS. Thus, the aim
of this study was to analyze the frequency of specific uncommon
chromosomal abnormalities in pMDS and their associations with the
clinical features, evolution from MDS to AML, and prognostic
significance.
Methods
Patients.
Cytogenetic and clinical studies were performed on 200 pediatric
patients with MDS between 2000 and 2022. Patients were diagnosed at
Instituto Nacional de Câncer, Instituto de Puericultura e Pediatria
Martagão Gesteira, and Hospital Universitário Oswaldo Cruz. Uncommon
chromosome alterations were present in 15 patients. These patients were
distributed according to sex as nine females and six males. The mean
age among these patients was five years old (ranging from 1 to 18
years). Most of the patients had pancytopenia with hypocellular bone
marrow (BM) showing dysplastic hematopoietic precursors and increased
blasts. None of these patients was previously treated for malignancy
nor had a previous diagnosis of genetic syndromes. The diagnosis and
classification were done according to the criteria proposed by the
International Consensus Classification of Myeloid Neoplasms and Acute
Leukemias.[1] The pediatric MDS patients were
classified as refractory cytopenia of childhood, RCC (5 patients),
MDS-EB (6 patients), and MDS/AML (4 patients).
Conventional and molecular cytogenetic analyses. The cytogenetic analysis from bone marrow cells was performed by G-banding as previously described by De Souza et al., 2014.[10] Chromosomes were identified and arranged according to the International System for Cytogenomic Nomenclature, 2020.[11]
Fluorescence in situ hybridization (FISH) analyses were done to confirm
the chromosome alterations using the following probes: D7S486 spectrum
orange/CEP7 spectrum Green, LSI MLL dual-color break-apart
rearrangement probe, LSI p53 spectrum orange, LSI EGR1 Spectrum Orange/
LSI D5S23, D5S721 Spectrum Green, LSI PML Spectrum Orange/ LSI RARA
Spectrum Green and LSI RUNX1T1 Spectrum Orange Probe/ RUNX1 Spectrum
Green Probe. The probes were from Vysis, Abbott Laboratories, USA. The
slides preparation was done according to manufacturer protocols.
Results
Uncommon
chromosome alterations were observed in 15 patients (7.5%) from 200
pMDS cases analyzed cytogenetically. These chromosome alterations were
divided into four cytogenetic groups: hyperdiploidy, uncommon
deletions, biclonal chromosomal alterations, and translocations, which
represented 33.3% (5/15), 13.3% (2/15), 33.3% (5/15), and 20% (3/15),
respectively. Of these patients, ten had disease progression before
HSCT, among them one received the HSCT and nine patients died during
disease evolution. Concerning the five patients who did not show
disease evolution, four underwent HSCT. Nowadays, only three patients
are still alive after HSCT. There is also a patient who remains stable
in the clinical course of MDS (Table 1).
 |
- Table
1.Pediatric MDS patients with uncommon cytogenetic alterations.
|
The
hyperdiploid karyotype represented 2.5% of all cases (5/200).
Hyperdiploidy was subdivided into two subgroups: patients who only had
chromosome gains (3/5) (Figure 1A)
and patients who also had structural alteration (2/5). The structural
alterations associated with hyperdiploid karyotype were: dup(1q);
der(6)del(6)(q21); der(12)del(12)(p11). Most of these patients had
advanced subtypes with severe pancytopenia and hypocellular BM. Three
patients had MDS-EB, one MDS/AML, and one with RCC. All these patients
were indicated for HSCT, but four had evolution to AML and three died
before the HSCT. Only two patients actually underwent HSCT, the
patient with RCC responded well to HSCT and is still alive. The patient
with hyperdiploid and structural alteration presented post-HSCT
cytogenetic and clinical relapse and died.
Deletions of the long arm of chromosome 5 were observed in two patients, one of them had also del(13q) (Figure 1B).
Both patients had pancytopenia and low blast count, being classified as
RCC and both were indicated for HSCT. Nevertheless, only one received
HSCT and this patient is still alive. The other patient did not have a
compatible donor and was treated with azacytidine, but unfortunately
died.
Biclonal karyotype was present in five patients,
representing 2.5% of all cases (5/200). In two patients, independent
clones, one with +8 and another with +21, were observed. Alterations
involving chromosome 7 were present in two patients: one patient with a
-7 clone and with a del(7q) clone (Figure 1 C-E).
The second patient had an add(7p) clone and +X and +8 clone. The other
patient had one clone with del(11)(q23) and another clone with
del(17)(p12), which evolved by acquiring a second alteration generating
a subclone del(17)(p12), del(12)(p13). Of patients with biclonal
chromosomal alterations, two had RCC, two had MDS-EB, and the other had
MDS/AML. All patients had severe pancytopenia and BM dysplasias. Four
patients were indicated for HSCT. However, two had disease progression
before HSCT and died. The follow-up post-HSCT showed disease relapse in
one patient who evolved to death, and the other patient is alive
without disease. The two patients with +8 and +21 are still waiting for
HSCT.
Chromosomal translocations were identified in three
patients, representing 1.5% of all cases (3/200). Two patients with
MDS/AML had balanced translocations, t(4;7)(p16;p15) and
t(5;8)(q32;q22). The t(4;7)(p16;p15) patient had pancytopenia whereas
the t(5;8)(q32;q22) patient had bicytopenia. In our study, unbalanced
translocation, der(2)t(2;15)(q37;q21), was also observed in one patient
with MDS-EB and normal cellularity. This patient also had three copies
of chromosome 15, two normal and one translocated to chromosome 2 (Figure 1 F, G). All patients had a progression to AML and died before the HSCT.
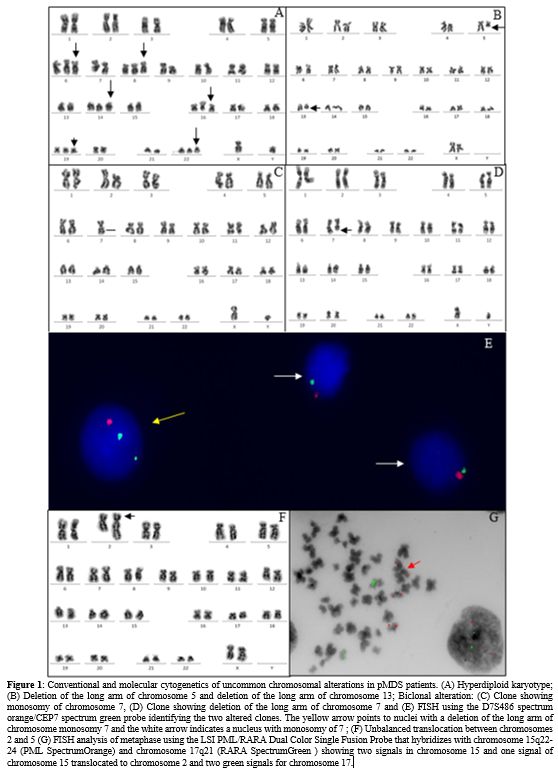 |
- Figure 1. Conventional
and molecular cytogenetics of uncommon chromosomal alterations in pMDS
patients. (A) Hyperdiploid karyotype; (B) Deletion of the long arm of
chromosome 5 and deletion of the long arm of chromosome 13; Biclonal
alteration: (C) Clone showing monosomy of chromosome 7, (D) Clone
showing deletion of the long arm of chromosome 7 and (E) FISH using the
D7S486 spectrum orange/CEP7 spectrum green probe identifying the two
altered clones. The yellow arrow points to nuclei with a deletion of
the long arm of chromosome monosomy 7 and the white arrow indicates a
nucleus with monosomy of 7 ; (F) Unbalanced translocation between
chromosomes 2 and 5 (G) FISH analysis of metaphase using the LSI
PML/RARA Dual Color Single Fusion Probe that hybridizes with chromosome
15q22-24 (PML SpectrumOrange) and chromosome 17q21 (RARA SpectrumGreen
) showing two signals in chromosome 15 and one signal of chromosome 15
translocated to chromosome 2 and two green signals for chromosome 17.
|
Discussion
Pediatric
MDS is characterized cytogenetically by clones containing alterations
that involve mainly chromosomal partial losses (deletions) or
chromosomal total losses (monosomies). These MDS cytogenetic patterns
suggest that this disease is associated mainly with the inactivation or
loss of tumor suppressor genes.[5,10]
Pediatric patients with acute lymphoblastic leukemia (ALL) present hyperdiploidy as a frequent cytogenetic abnormality.[12,13]
Hyperdiploidy can be divided into two main subtypes: high hyperdiploidy
(51-65 chromosomes) associated with favorable prognosis, and low
hyperdiploidy (47-50 chromosomes) related with unfavorable prognosis.[12,13]
In MDS, the hyperdiploid karyotype is rare. However, patients with MDS
may have complex karyotypes, with three or more chromosomal
alterations, and are classified as very poor prognosis according to the
IPSS-R.[3] So, it is important to note the presence of hyperdiploidy in these cases.
Previously, our group reported the first case of high hyperdiploid karyotype in pMDS.[14]
In this study, we describe more four cases of hyperdiploid karyotype.
However, these patients showed low hyperdiploidy. Three patients
presented structural alterations such as deletions and duplications,
which is uncommon in hyperdiploid karyotypes.[13] The
hyperdiploid was observed in the initial subtype and advanced subtypes.
However, the HSCT was successful only in the initial subtype,
highlighting the importance of an early diagnosis and indication for
this treatment.
The most frequent cytogenetic alterations in
adult MDS is del(5q), which is associated generally with a favorable
prognosis and defines a unique MDS sub-category.[1,15] Nevertheless, del(5q) is extremely rare in children, and it seems to be associated with poor outcomes.[16] In our study, the del(5q) was observed isolated, as previously published[17]
and with del(13q). Both patients were indicated for HSCT. The patient
with del(5q) as sole chromosomal abnormality had a good outcome
post-transplant. Nevertheless, the other patient did not have a
compatible donor. This patient was treated with azacytidine but showed
disease evolution and died. Although it has been demonstrated that
azacitidine is an efficient and safe MDS therapy for adult patients,
data for this treatment in children is still lacking. In children,
there is no established treatment to prevent or delay progression to
leukemia before HSCT. However, some studies have shown that azacitidine
is effective in some children with MDS and appears to be a non-toxic
option in palliative situations to prolong survival.[18,19]
Another
uncommon finding in our study was unrelated clones, also known as
biclonal chromosomal alterations, detected in one sample simultaneously
by G-banding analysis. There are different hypotheses about the
mechanisms that lead to these alterations. However, the actual
mechanism is still unknown. Some authors believe those unrelated clones
have the same founding molecular mutation and acquire different
alterations over evolution, thus giving rise to unrelated cytogenetic
clones.[20-23] Nevertheless, nowadays there are
molecular models of MDS development showing that distinct stem cells
had different genetic variants at the same time.[1]
In
adult patients, biclonal chromosomal alterations are also categorized
as rare chromosomal abnormalities, representing 4.3-6.7% of the cases
and being associated with disease relapse. These studies showed that
the most recurrent chromosome alterations in unrelated clones were
del(5q), +8, del(20q), del(7q), +11, +21, and -22.[20,21] Previously, our group reported the first case of biclonal chromosomal alteration in a pMDS.[22]
The present study observed a frequency of biclonal chromosomal
abnormalities of 2.5% (5/200), involving +8 and +21 as the most
recurrent alterations. The leukemic evolution was observed in two
patients (2/5), but it is important to note that the others were
treated with HSCT. Furthermore, two patients after HSCT had cytogenetic
relapse and death, showing how difficult it is to treat patients with
such chromosomal instability.
In pMDS, chromosomal translocations are uncommon findings and associated with unfavorable prognosis.[24]
In this study, two patients had balanced chromosomal translocation:
t(5;8)(q32;q22), t(4;7)(p16;p15), and one patient showed unbalanced
translocation der(2)t(2;15)(q37;q21). These alterations were not
previously reported in hematological neoplasm according to the Atlas of
Genetics and Cytogenetics in Oncology and Haematology, 2022.[15]
The patients with balanced and unbalanced translocations were diagnosed
with MDS/AML, showed disease progression to AML and died. The
unfavorable outcome of our patients suggests that the chromosomal
translocations are associated with an adverse prognosis.
Chromosomal
abnormalities play an essential role in the diagnosis and prognosis of
patients with MDS, but approximately 50% of patients have a normal
karyotype observed by G-banding analysis. In these cases, complementary
molecular methodologies may provide relevant prognostic information,
such as the analysis using next-generation sequencing (NGS).[25]
Identification of genetic variants through the NGS opens new
opportunities to characterize the genomic architecture of patients with
MDS and contributes to the establishment of prognostic biomarkers.[25-28]
In this sense, it was developed the Molecular-IPSS for adult patients,
which integrates the cytogenetic, molecular, and hematological
features.[28] In our study, although the focus was on
cytogenetics, the analysis using NGS with a customized panel could
provide complementary information associated with the prognosis
reinforcing our findings. However, due to the high cost of NGS tests,
these are not yet a reality globally used, mainly in developing
countries. So, cytogenetics continues to play an important role for
patients with hematologic malignancies, mainly for pMDS where yet
little is known about the predictive value for molecular alterations
due to the rarity of this disease. Since this study was the first with
a large cohort of patients with pMDS focusing on rare chromosomal
alterations and their impact on prognosis, it is necessary to confirm
our results in other cohorts to provide a better understanding and to
determine the true prognostic value of these uncommon chromosomal
alterations in pMDS.
Conclusions
In
summary, in our study, the uncommon chromosomal alterations in pMDS
were associated with unfavorable prognosis. The study of uncommon
cytogenetic alterations in pMDS is extremely important to contribute to
the stratification of cytogenetic risk groups and early indication of
HSCT.
Acknowledgements
This
study was supported by Fundação Carlos Chagas Filho de Amaro à Pesquisa
do Estado do Rio de Janeiro (FAPERJ) (FAPERJ/E-26/201.2018/2022) and
the Brazilian Ministry of Health (Instituto Nacional de Câncer/INCA,
Brazil).
Author Contributions
VLL,
BFS, and TSF wrote the manuscript. TSF designed the study. VLL, BFS,
EFR, MLRRB, and TJMS performed the cytogenetic and FISH analysis. RCBT,
APB, and ESC analyzed the clinical data. TSF and TJMS reviewed
critically the manuscript for important intellectual content. All
authors have read and approved the manuscript.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding Statement
This
study was supported by Fundação Carlos Chagas Filho de Amaro à Pesquisa
do Estado do Rio de Janeiro (FAPERJ) (FAPERJ/E-26/201.2018/2022) and
the Brazilian Ministry of Health (Instituto Nacional de Câncer/INCA,
Brazil).
Ethics Approval and Consent to Partecipate
This
study was approved by the Ethics and Research Committee of the National
Cancer Institute (reference number # 3401739) in accordance with the
Declaration of Helsinki. Informed consent was obtained from the
children’s parents.
References
- Arber DA, Orazi A, Hasserjian RP, et al.
International Consensus Classification of Myeloid Neoplasms and Acute
Leukemias: integrating morphologic, clinical, and genomic data. Blood.
2022;140(11):1200-1228. https://doi.org/10.1182/blood.2022015850 PMid:35767897 PMCid:PMC9479031
- Hasle
H. Myelodysplastic and myeloproliferative disorders of childhood.
Hematology Am Soc Hematol Educ Program. 2016; 2016(1):598-604. https://doi.org/10.1182/asheducation-2016.1.598 PMid:27913534 PMCid:PMC6142519
- Greenberg
PL, Tuechler H, Schanz J, et al. Revised international prognostic
scoring system for myelodysplastic syndromes. Blood.
2012;120(12):2454-65. https://doi.org/10.1182/blood-2012-03-420489 PMid:22740453 PMCid:PMC4425443
- Moriwaki
K, Manabe A, Taketani T, et al. Cytogenetics and clinical features of
pediatric myelodysplastic syndrome in Japan. Int J Hematol.
2014;100(5):478-84. https://doi.org/10.1007/s12185-014-1674-z PMid:25261124
- Bacher
U, Schanz J, Braulke F, Haase D. Rare cytogenetic abnormalities in
myelodysplastic syndromes. Mediterr J Hematol Infect Dis. 2015;
7:e2015034. https://doi.org/10.4084/mjhid.2015.034 PMid:25960862 PMCid:PMC4418404
- Lamim
Lovatel V, Otero L, Orlando EP, et al. Clinical and Prognostic Features
in a Young Adult Patient with de novo Myelodysplastic Syndrome
Presenting t(11;16)(q23;q24). Mediterr J Hematol Infect Dis.
2022;14(1):e2022013. https://doi.org/10.4084/MJHID.2022.013 PMid:35070220 PMCid:PMC8747008
- Grimwade
D, Hills RK, Moorman AV, et al. Refinement of cytogenetic
classification in acute myeloid leukemia: determination of prognostic
significance of rare recurring chromosomal abnormalities among 5876
younger adult patients treated in the United Kingdom Medical Research
Council trials. Blood. 2010;116(3):354-65. https://doi.org/10.1182/blood-2009-11-254441 PMid:20385793
- Mrózek
K, Kohlschmidt J, Blachly JS, et al. Outcome prediction by the 2022
European LeukemiaNet genetic-risk classification for adults with acute
myeloid leukemia: An Alliance study. Leukemia. 2023;37(4):788-798. https://doi.org/10.1038/s41375-023-01846-8 PMid:36823396 PMCid:PMC10079544
- Platte
V, Bergmann A, Hildebrandt B, et al. Clinical and Cytogenetic
Characterization of Early and Late Relapses in Patients Allografted for
Myeloid Neoplasms with a Myelodysplastic Component. Cancers.
2022;14(24):6244. https://doi.org/10.3390/cancers14246244 PMid:36551729 PMCid:PMC9776604
- de
Souza DC, Fernandez Cde S, Camargo A, et al. Cytogenetic as an
important tool for diagnosis and prognosis for patients with
hypocellular primary myelodysplastic syndrome. Biomed Res Int.
2014;2014:542395. https://doi.org/10.1155/2014/542395 PMid:25180186 PMCid:PMC4144075
- McGowan-Jordan J, Hastings RJ, Moore S.ISCN 2020: an International System for Human Cytogenomic Nomenclature. S Karger AG;2020. https://doi.org/10.1159/isbn.978-3-318-06867-2 PMid:31568709
- Enshaei
A, Vora A, Harrison CJ, et al. Defining low-risk high hyperdiploidy in
patients with pediatric acute lymphoblastic leukaemia: a retrospective
analysis of data from the UKALL97/99 and UKALL2003 clinical trials.
Lancet Haematol. 2021;8(11):e828-e839. https://doi.org/10.1016/S2352-3026(21)00304-5 PMid:34715050
- Lejman
M, Chałupnik A, Chilimoniuk Z, Dobosz M. Genetic Biomarkers and Their
Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in
Children. Int J Mol Sci. 2022;23(5):2755. https://doi.org/10.3390/ijms23052755 PMid:35269896 PMCid:PMC8911213
- de
Souza Fernandez T, Ornellas MH, Tavares Rde C, et al. Hyperdiploid
karyotype in a child with hypocellular primary myelodysplastic
syndrome. Eur J Haematol. 2003;71(5):399-401. https://doi.org/10.1034/j.1600-0609.2003.00150.x PMid:14667207
- Atlas
of Genetics and Cytogenetics in Oncology and Haematology in 2013. Huret
JL, Ahmad M, Arsaban M, et al. Nucleic Acids Res. 2013 Jan;41(Database
issue):D920-4. https://doi.org/10.1093/nar/gks1082 PMid:23161685 PMCid:PMC3531131
- Gurnari
C, Piciocchi A, Soddu S. et al. Myelodysplastic syndromes with del(5q):
A real-life study of determinants of long-term outcomes and response to
lenalidomide. Blood Cancer J. 2022; 12(9):132. https://doi.org/10.1038/s41408-022-00724-3 PMid:36071048 PMCid:PMC9452671
- de
Souza DC, Otero L, Tavares R de C, et al. An uncommon case of a child
with del(5q) and hypocellular myelodysplastic syndrome. Pediatr Blood
Cancer. 2010;55(4):767. https://doi.org/10.1002/pbc.22633 PMid:20589660
- Cseh
AM, Niemeyer CM, Yoshimi A, et al. Therapy with low-dose azacitidine
for MDS in children and young adults: a retrospective analysis of the
EWOG-MDS study group. Br J Haematol. 2016;172(6):930-936. https://doi.org/10.1111/bjh.13915 PMid:26766110
- Waespe
N, Van Den Akker M, Klaassen RJ, et al. Response to treatment with
azacitidine in children with advanced myelodysplastic syndrome prior to
hematopoietic stem cell transplantation. Haematologica
2016;101(12):1508-1515. https://doi.org/10.3324/haematol.2016.145821 PMid:27540140 PMCid:PMC5479623
- Han
JY, Theil KS, Hoeltge G. Frequencies and characterization of
cytogenetically unrelated clones in various hematologic malignancies:
seven years of experiences in a single institution. Cancer Genet
Cytogenet. 2006;164(2):128-32. https://doi.org/10.1016/j.cancergencyto.2005.07.013 PMid:16434315
- Han
JY, Kim KH, Kwon HC, et al. Unrelated clonal chromosome abnormalities
in myelodysplastic syndromes and acute myeloid leukemias. Cancer Genet
Cytogenet. 2002;132(2):156-8. https://doi.org/10.1016/S0165-4608(01)00549-0 PMid:11850080
- Rodrigues
EF, de Souza DC, Camargo A, et al. Cytogenetic biclonality in a child
with hypocellular primary myelodysplastic syndrome. Cancer Genet
Cytogenet. 2007; 178(1):70-2. https://doi.org/10.1016/j.cancergencyto.2007.05.025 PMid:17889712
- Chen
J, Kao YR, Sun D, et al. Myelodysplastic syndrome progression to acute
myeloid leukemia at the stem cell level. Nat Med. 2019; 25(1):103-110. https://doi.org/10.1038/s41591-018-0267-4 PMid:30510255 PMCid:PMC6436966
- Koppalkar
RK, Rao PS, Sandhya I, Muktha R Pai. A Rare Translocation in a
Paediatric Myelodysplastic Syndrome. Journal of Clinical and Diagnostic
Research. 2018;12(12): ED07-ED09. https://doi.org/10.7860/JCDR/2018/37527.12330
- Zeng
X, Zhang Y, Zhao K, et al. Somatic mutations predict prognosis in
myelodysplastic syndrome patients with normal karyotypes. Signal
Transduct Target Ther. 2021;6(1):274. https://doi.org/10.1038/s41392-021-00606-3 PMid:34305138 PMCid:PMC8310889
- Xu
L, Gu ZH, Li Y, et al. Genomic landscape of CD34+ hematopoietic cells
in myelodysplastic syndrome and gene mutation profiles as prognostic
markers. Proc Natl Acad Sci U S A. 2014;111(23):8589-94. https://doi.org/10.1073/pnas.1407688111 PMid:24850867 PMCid:PMC4060725
- Schwartz
JR, Ma J, Lamprecht T, et al. The genomic landscape of pediatric
myelodysplastic syndromes. Nat Commun. 2017;8(1):1557. https://doi.org/10.1038/s41467-017-01590-5 PMid:29146900 PMCid:PMC5691144
- Bernard
E, Tuechler H, Greenberg PL, et al. Molecular international prognostic
scoring system for myelodysplastic syndromes. NEJM evidence, 2022;1(7),
EVIDoa 2200008.