Materials and Methods
In
this retrospective study, 52 consecutive SCD patients who were
diagnosed as Hemoglobin S/D Punjab Heterozygotes between 2006 and 2022
at the Sultan Qaboos University Hospital in Oman were enrolled,
following written approval from the local medical research and ethics
committee (MREC # 2551). The data was obtained from the electronic
patient record system and included demographic features SCD related
manifestations such as the frequency of painful vasoocclusive crises,
ACS, splenic sequestration, AVN, priapism, gallstones and Stroke. We
also analyzed the haematological, biochemical and radiological
parameters, as well as the use of hydroxyurea therapy. The laboratory
results obtained included complete blood counts, haemoglobin
electrophoresis data, and alpha thalassemia status (when available).
Biochemical parameters included renal and liver function tests along
with serum LDH levels. We utilized data from historical controls of our
previous studies relating to other genotypes of SCD in our population.[12]
Statistical Analysis. The statistical package for social science (IBM SPSS, USA ver.23, Armonk, NY) was used to analyze the collected data. Normally distributed data was characterized as mean with standard deviation, whereas data that was not normally distributed was characterized as median with interquartile range (IQR) for continuous variables and percentage and frequency for categorical variables. Clinical and laboratory parameters were compared using the means for continuous measures and tested for association using the Student t-test, Fisher’s exact test and chi-square test. A P value of <0.05 was considered to be statistically significant.
Statistical Analysis. The statistical package for social science (IBM SPSS, USA ver.23, Armonk, NY) was used to analyze the collected data. Normally distributed data was characterized as mean with standard deviation, whereas data that was not normally distributed was characterized as median with interquartile range (IQR) for continuous variables and percentage and frequency for categorical variables. Clinical and laboratory parameters were compared using the means for continuous measures and tested for association using the Student t-test, Fisher’s exact test and chi-square test. A P value of <0.05 was considered to be statistically significant.
Results
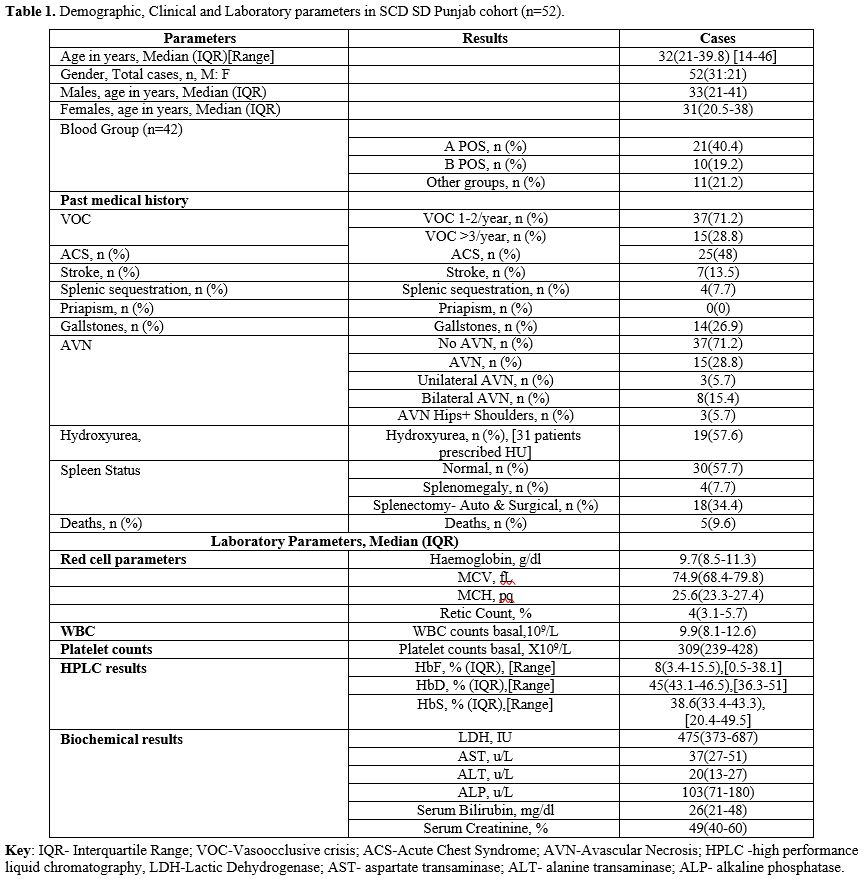
Amongst
the 52 SCD SD Punjab double heterozygotes, 31 (59.6%) were males with a
median age (IQR) of 33 (21-41) years and a range between 14 to 46 years
(Table 1). 31% of this cohort
were below 25 years old, whereas the age of majority (69%) ranged
between 25 to 46 years. There was no gender preponderance for the SCD
manifestations like VOC events, ACS, splenic sequestration or Stroke
and mortality in this cohort, but splenic atrophy/splenectomy was more
common in females (p=0.007, Fisher’s exact test).
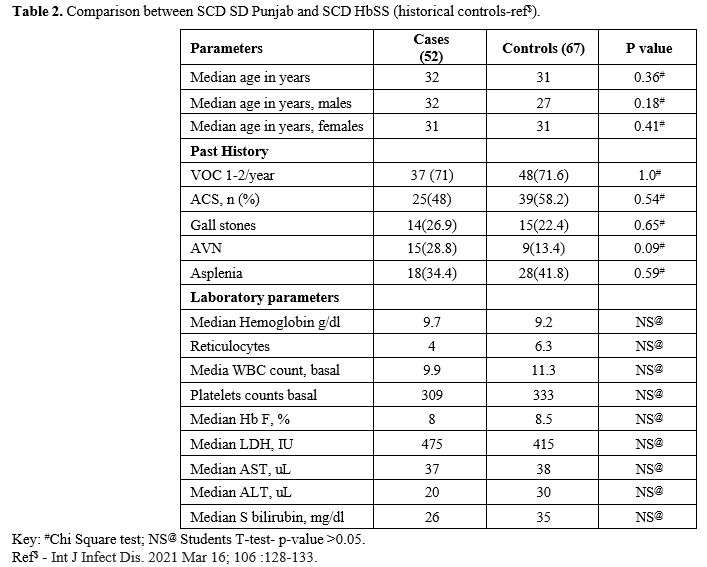
Table 2 shows no significant differences when a historical cohort of other SCD genotype patients that are followed at our centre were compared with our current cohort of SD SCD patients. These are not head-to-head comparisons but historical data of other SCD genotype patients from previous publications.
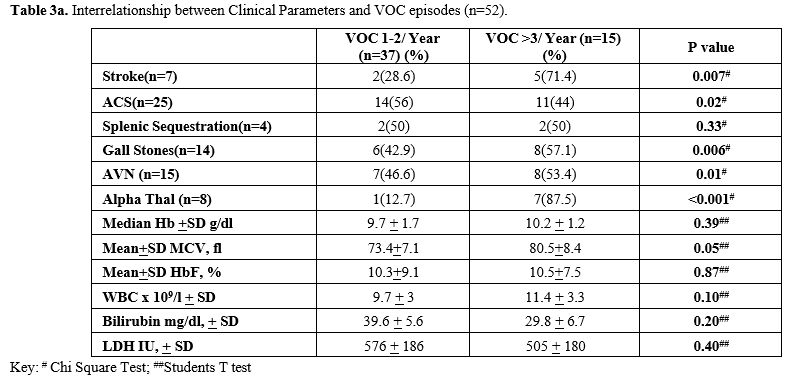
Recurrent VOC events were recorded in all the patients, with 71.2% showing less than 3 episodes per year and 28.8% showing 3 or more episodes per year. Many of these patients had a number of acute sickle cell- related complications, including ACS, Stroke, and splenic sequestration in 48%, 13.5% and 7.7%, respectively. Stroke was more common in the patients with VOC episodes >3/year (p=0.007, Chi-square test). On the contrary, ACS was more frequent in patients with VOC episodes <3/year, but this association was not statistically significant (p=0.02, Chi-square test) (Table 3a).
Further, chronic complications such as AVN and gallstones were seen in 28.8% and 26.9%, respectively. AVN and Gall stones were more prevalent in patients with VOC episodes > 3/year, and this association was statistically significant (p=0.01 and p=0.006, respectively, Chi-square test). Most patients had normal spleen (57.7%), however, absent spleen was confirmed in 34.6% (Auto or surgical splenectomy), whereas 7.7% had splenomegaly. Further, there was a significant association of splenic sequestration with splenectomy (p=0.002, Fisher's Exact test). Lastly, although 59.6% were males, no case of priapism was reported in this cohort. The most common blood group was A+ (40.4%), followed by B+ (19.2%), O+ (13.4%), O- (5.7%), and AB+ (1.9%).
Amongst the haematology parameters, the median (IQR) Hb (g/dl), MCV (fl), Retic count (%), WBC count (X109/L) and Platelet count (X109/L) were 9.7(8.5-11.3), 74.9 (68.4-79.8), 4 (3.1-5.7), 9.9 (8.1-12.6) and 309 (239-428) respectively. However, leukocytosis and thrombocytosis were present in 32% and 26% of this SD- Punjab cohort, respectively, with thrombocytopenia present in 10% of the group.
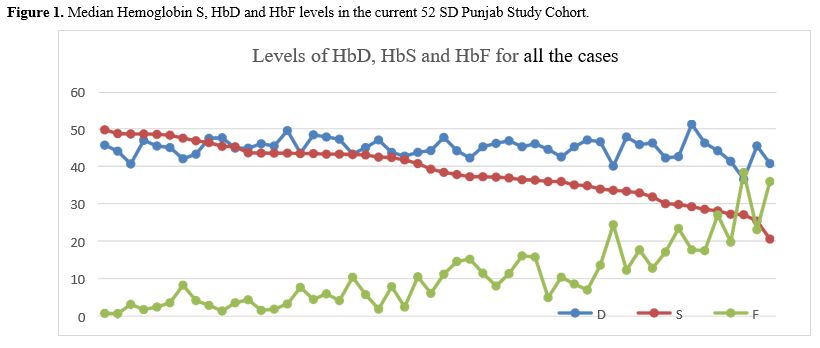
Haemoglobin variant analysis revealed that haemoglobin S and haemoglobin D were the two prominent variants in these Hb SD Punjab patients. Further, it was observed that as the HbF fraction increased, the level of HbS decreased in these patients. Additionally, data on the BS genotype, Alpha genotype, and Sickle Haplotype details were available, albeit in a small proportion of patients in this cohort only.
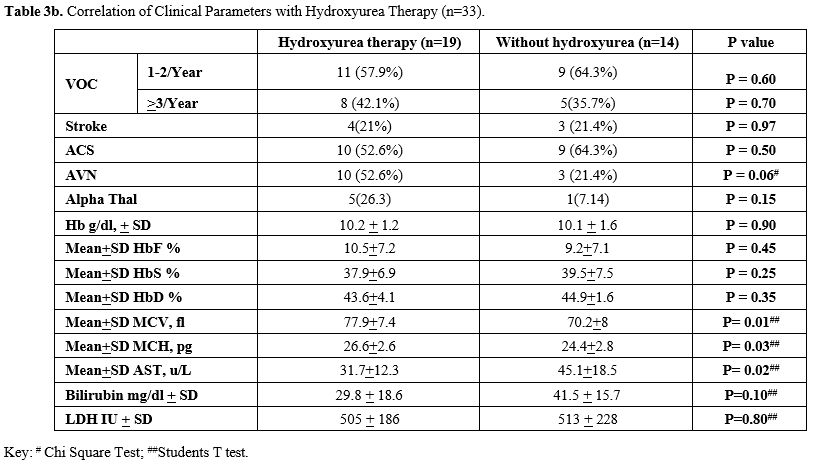
The biochemical analysis of liver function tests, creatinine and LDH were found to correlate with disease activity, with the majority showing normal liver functions, but raised LDH and serum bilirubin levels and low serum creatinine levels. The interrelationship severity of vasoocclusive crisis and outcome parameters is reported in Table 3a, which shows comparable levels of Hb and also HbF, as well as WBC, between those with mild disease vs. moderate to severe. Similarly, there were statistically significant differences in patients with Stroke, gall stones, presence of alpha thalassaemia and AVN (Chi-Square test). The study did not find any statistically significant association between HU therapy and VOC episodes, ACS, Splenic sequestration, AVN, Gall stone, Stroke or Mortality (Table 3b). However, there was a statistically significant difference in patients who took HU with a higher mean MCV and MCH values (Students t-test).
 |
|
Table 2 shows no significant differences when a historical cohort of other SCD genotype patients that are followed at our centre were compared with our current cohort of SD SCD patients. These are not head-to-head comparisons but historical data of other SCD genotype patients from previous publications.
 |
|
Recurrent VOC events were recorded in all the patients, with 71.2% showing less than 3 episodes per year and 28.8% showing 3 or more episodes per year. Many of these patients had a number of acute sickle cell- related complications, including ACS, Stroke, and splenic sequestration in 48%, 13.5% and 7.7%, respectively. Stroke was more common in the patients with VOC episodes >3/year (p=0.007, Chi-square test). On the contrary, ACS was more frequent in patients with VOC episodes <3/year, but this association was not statistically significant (p=0.02, Chi-square test) (Table 3a).
 |
|
Further, chronic complications such as AVN and gallstones were seen in 28.8% and 26.9%, respectively. AVN and Gall stones were more prevalent in patients with VOC episodes > 3/year, and this association was statistically significant (p=0.01 and p=0.006, respectively, Chi-square test). Most patients had normal spleen (57.7%), however, absent spleen was confirmed in 34.6% (Auto or surgical splenectomy), whereas 7.7% had splenomegaly. Further, there was a significant association of splenic sequestration with splenectomy (p=0.002, Fisher's Exact test). Lastly, although 59.6% were males, no case of priapism was reported in this cohort. The most common blood group was A+ (40.4%), followed by B+ (19.2%), O+ (13.4%), O- (5.7%), and AB+ (1.9%).
Amongst the haematology parameters, the median (IQR) Hb (g/dl), MCV (fl), Retic count (%), WBC count (X109/L) and Platelet count (X109/L) were 9.7(8.5-11.3), 74.9 (68.4-79.8), 4 (3.1-5.7), 9.9 (8.1-12.6) and 309 (239-428) respectively. However, leukocytosis and thrombocytosis were present in 32% and 26% of this SD- Punjab cohort, respectively, with thrombocytopenia present in 10% of the group.
Haemoglobin variant analysis revealed that haemoglobin S and haemoglobin D were the two prominent variants in these Hb SD Punjab patients. Further, it was observed that as the HbF fraction increased, the level of HbS decreased in these patients. Additionally, data on the BS genotype, Alpha genotype, and Sickle Haplotype details were available, albeit in a small proportion of patients in this cohort only.
The biochemical analysis of liver function tests, creatinine and LDH were found to correlate with disease activity, with the majority showing normal liver functions, but raised LDH and serum bilirubin levels and low serum creatinine levels. The interrelationship severity of vasoocclusive crisis and outcome parameters is reported in Table 3a, which shows comparable levels of Hb and also HbF, as well as WBC, between those with mild disease vs. moderate to severe. Similarly, there were statistically significant differences in patients with Stroke, gall stones, presence of alpha thalassaemia and AVN (Chi-Square test). The study did not find any statistically significant association between HU therapy and VOC episodes, ACS, Splenic sequestration, AVN, Gall stone, Stroke or Mortality (Table 3b). However, there was a statistically significant difference in patients who took HU with a higher mean MCV and MCH values (Students t-test).
 |
|
Discussion
Compound
heterozygosity of HbS and other haemoglobin variants like HbC, HbE, and
HbD gives a great clinical and haematological heterogeneity within SCD4. Although HbS/D Punjab has a multicentric origin, it is highly prevalent in the Punjab region of India and Pakistan.[8] Hemoglobin-D does not cause sickling but has an electrophoretic mobility indistinguishable from that of hemoglobin-S.[13]
Although Hb D trait and Homozygous D are relatively asymptomatic, with
no underlying haemolytic anaemia, in compound heterozygous Hb SD Punjab
disease, Hb D Punjab interacts with Hb S to enhance polymerization and
sickling process and produces moderately severe sickle cell disease
phenotype and hemolysis.[10] SCD is a heterogeneous
disease with variable clinical and laboratory expression due to
multiple genetic and environmental factors.[14]
Adekile A. et al. in 2010 reported that Hb-SD disease in patients from
Kuwait was associated with mild to moderate splenomegaly, recurrent
hemolytic anaemia, acute chest syndrome, and also AVN of the hips.[9]
Further, Al-Barazanchi et al., in their report on 42 patients from
Iraq, observed that recurrent vaso-occlusive events were similar to
sickle cell anaemia, and the clinical course was indistinguishable from
homozygous Hb SS.[10] In keeping with the above
literature, we evaluated 52 patients with SD, finding that recurrent
VOC events were seen in all these patients, with moderate to severe VOC
episodes (>3/year) that were seen in about a third of this cohort
(28.8%). Among the group with severe recurrent VOC (>3 /ear), we
found them more likely to have Stroke, Gall stones and AVN, although
the level of Hb F was similar in both groups (Table 3a).
Also, our cohort showed significant other complications, including ACS
in 48%, Stroke in 13.5% and splenic sequestration in 7.7%. Among the
chronic complications of SD, AVN and gallstones were present in 28.8%
and 26.9%, respectively. These findings were similar to those of other
SCD genotypes within our community, as indicated in Table 2.
The impact of HbF is not clear and raises the question about the lack
of protective effect in this group of patients, a point that was also
raised previously.[9] Similarly, among the AVN group,
hip joints were the most affected, but shoulders were also involved.
Also, these patients have lost their spleen (surgical and
autosplenectomy) in about 35% of patients, which is similar to reports
in other SCD genotypes from our region.[15]
The laboratory data showed that the Hb Level, WBC, and platelets were similar to what is expected in patients with SS and Sβo with normal renal and hepatic functions. Also, patients showed a relatively high HbF generally, with and without hydroxyurea, and a decline in Hb S level, with rising HbF, with the Hb D level remaining constant.
Further, the laboratory findings reveal a basal Hb for this cohort of 9.7 g/dl, which is generally similar to what is expected for other SCD groups. Values for WBC and platelets are also as expected. However, basal HbF is relatively high at 8%, and it was as high as 38% in some cases. Amongst the 33 patients on HU therapy, as expected, patients had a higher MCV & MCH (statistically significant), with higher Hb F; however, it did not have a significant impact on the frequency of VOC, Stroke or ACS. Although there is a trend towards the development of AVN, it did not reach statistical significance and justify the earlier comment about the protective role of hydroxyurea and HbF in SD-related complications (Table 3b). Interestingly, among the 19 patients on HU, 4 patients (21%) had Stroke. Further, although 57.6% were taking HU, ACS was seen in a proportionately higher number of patients (75%) than those who did not take HU (53%). However, this difference also was not statistically significant. Furthermore, the subset of patients with increased severity in the HU group, represented by >3 VOCs/year, had a significantly higher MCV and MCH, indicative not only of an effect of HU, but also of a probable role of the high incidence of alpha thalassaemia, that has been reported in the Omani population and is known to ameliorate the severity of VOC’s.[2,16,17]
In contrast to the HbSS patients who with a high HbF had a milder clinical severity Haemoglobin SD compound heterozygotes from Kuwait with a higher HbF demonstrated a severe clinical course.[9] This reflected the high prevalence of Arab-Indian haplotype in the Kuwaiti SCD population.[9] Further, it also appeared that the interaction of HbD and HbF was unable to prevent or inhibit the polymerization of HbS in spite of the high HbF levels, nor did it ameliorate the clinical course of SCD in this population. In our cohort, although the IQR of HbF ranged from 3.4-15.5% with a median of 8%, the actual range of the literature cases was from 0.5% to 38.1%. This also reflects the mixed SCD haplotypes of Benin, Bantu, and Arab-Indian reported in our SCD population.[18] In the small subset of these patients where data of the sickle haplotypes were available, the mean HbF (SD) in the Bantu haplotype was 1.8+1.1, with only one-third of patients on HU, while, in the Arab Indian haplotype, the mean (SD) was 13.4+5.7 with two-thirds of the patients taking HU, whereas, in the Benin haplotype, the mean HbF (SD) was 13.5+3.3 with all the subgroup patients taking HU. Thus, since the number of subjects was small, any further statistical analysis /conclusions were not possible. Nevertheless, this study did not find any significant association between HU therapy and VOC episodes, ACS, Splenic sequestration, AVN, Gall stone, Stroke or mortality.
Interestingly, we found that the most common blood group in this cohort was A+(40.4%), whereas the most recent study from the country revealed that A+ was only seen in 17.4%, while the most common blood group reported from Oman was O+ (44.9%) followed by B+ (20.2%), A+ (17.4%), AB+ (6.8%), B- (2.7%), O- (7.4%), and A- (0.6%).[18] It is difficult to explain this observation, and a larger sample size is needed to make comparative observations. Nevertheless, a founder effect may explain this observation.
The laboratory data showed that the Hb Level, WBC, and platelets were similar to what is expected in patients with SS and Sβo with normal renal and hepatic functions. Also, patients showed a relatively high HbF generally, with and without hydroxyurea, and a decline in Hb S level, with rising HbF, with the Hb D level remaining constant.
 |
|
Further, the laboratory findings reveal a basal Hb for this cohort of 9.7 g/dl, which is generally similar to what is expected for other SCD groups. Values for WBC and platelets are also as expected. However, basal HbF is relatively high at 8%, and it was as high as 38% in some cases. Amongst the 33 patients on HU therapy, as expected, patients had a higher MCV & MCH (statistically significant), with higher Hb F; however, it did not have a significant impact on the frequency of VOC, Stroke or ACS. Although there is a trend towards the development of AVN, it did not reach statistical significance and justify the earlier comment about the protective role of hydroxyurea and HbF in SD-related complications (Table 3b). Interestingly, among the 19 patients on HU, 4 patients (21%) had Stroke. Further, although 57.6% were taking HU, ACS was seen in a proportionately higher number of patients (75%) than those who did not take HU (53%). However, this difference also was not statistically significant. Furthermore, the subset of patients with increased severity in the HU group, represented by >3 VOCs/year, had a significantly higher MCV and MCH, indicative not only of an effect of HU, but also of a probable role of the high incidence of alpha thalassaemia, that has been reported in the Omani population and is known to ameliorate the severity of VOC’s.[2,16,17]
In contrast to the HbSS patients who with a high HbF had a milder clinical severity Haemoglobin SD compound heterozygotes from Kuwait with a higher HbF demonstrated a severe clinical course.[9] This reflected the high prevalence of Arab-Indian haplotype in the Kuwaiti SCD population.[9] Further, it also appeared that the interaction of HbD and HbF was unable to prevent or inhibit the polymerization of HbS in spite of the high HbF levels, nor did it ameliorate the clinical course of SCD in this population. In our cohort, although the IQR of HbF ranged from 3.4-15.5% with a median of 8%, the actual range of the literature cases was from 0.5% to 38.1%. This also reflects the mixed SCD haplotypes of Benin, Bantu, and Arab-Indian reported in our SCD population.[18] In the small subset of these patients where data of the sickle haplotypes were available, the mean HbF (SD) in the Bantu haplotype was 1.8+1.1, with only one-third of patients on HU, while, in the Arab Indian haplotype, the mean (SD) was 13.4+5.7 with two-thirds of the patients taking HU, whereas, in the Benin haplotype, the mean HbF (SD) was 13.5+3.3 with all the subgroup patients taking HU. Thus, since the number of subjects was small, any further statistical analysis /conclusions were not possible. Nevertheless, this study did not find any significant association between HU therapy and VOC episodes, ACS, Splenic sequestration, AVN, Gall stone, Stroke or mortality.
Interestingly, we found that the most common blood group in this cohort was A+(40.4%), whereas the most recent study from the country revealed that A+ was only seen in 17.4%, while the most common blood group reported from Oman was O+ (44.9%) followed by B+ (20.2%), A+ (17.4%), AB+ (6.8%), B- (2.7%), O- (7.4%), and A- (0.6%).[18] It is difficult to explain this observation, and a larger sample size is needed to make comparative observations. Nevertheless, a founder effect may explain this observation.
Conclusions
Although
this is a retrospective study of the SCD-SD Punjab genotype, it is one
of the largest single-centre studies. It demonstrated that SD disease
is associated with significant morbidity and mortality, similar to
other SCD genotypes. It also demonstrated that although Hb F is high,
and with the use of HU, its impact on disease morbidity and mortality
needs to be clarified, and longer follow-up on a larger sample of these
patients is needed. Although this study is the largest single-centre
study of the SCD SD genotype, however, it is a retrospective study, and
the sample size is relatively small. Thus, more collaborative
prospective work is needed to define the precise clinical and
laboratory features of this specific SCD genotype.
Acknowledgements
We wish to thank the hospital administration for the use of hospital material in this study.
Author Contributions
All
authors have made substantial contributions, have seen, and approved
the final version of manuscript. SAK and AVP were fully involved in the
conception and design of the study, recruitment and care of patients,
acquisition of data, analysis and interpretation of data and was
instrumental in the drafting the article and critical appraisal before
submission.
References
- Colombatti R, Birkegard C, Medici M, PB2215:
Global epidemiology of sickle cell disease: A systematic review.
HemaSphere 6: p 2085-2086, June 2022. https://doi.org/10.1097/01.HS9.0000851688.00394.f4 PMCid:PMC9431667
- Alkindi
S, Al Zadjali S, Al Madhani A, Daar S, Al Haddabi H, Al Abri Q, et al.
Forecasting Hemoglobinopathy Burden Through Neonatal Screening in Omani
Neonates, Hemoglobin. 2010; 34(2), 135-144. https://doi.org/10.3109/03630261003677213 PMid:20353348
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017 Jul 15; 390 (10091):311-323. https://doi.org/10.1016/S0140-6736(17)30193-9 PMid:28159390
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997 Sep 11;337(11):762-9. https://doi.org/10.1056/NEJM199709113371107 PMid:9287233
- Savitt
TL, Goldberg MF. Herrick's 1910 case report of sickle cell anemia. The
rest of the story. JAMA. 1989 Jan 13;261(2):266-71. https://doi.org/10.1001/jama.261.2.266 PMid:2642320
- Yavarian
M, Karimi M, Paran F, Neven C, Harteveld CL, Giordano PC,Multi centric
origin of Hb D-Punjab [beta121(GH4)Glu-->Gln, GAA>CAA],
Hemoglobin 2009;33 (6): 399-405. https://doi.org/10.3109/03630260903344598 PMid:19958184
- Atalay
EO, Atalay A, Ustel E, Yildiz S, Oztürk O, Köseler A, Bahadir A,
Genetic origin of Hb D-Los Angeles [beta121(GH4) Glu-->Gln,
GAA-->CAA] according to the beta-globin gene cluster haplotypes,
Hemoglobin. 2007;31(3):387-91. https://doi.org/10.1080/03630260701459416 PMid:17654078
- Patel
DK, Mashon RS, Patel S, Dash PM, Das BS, β-globin gene haplotypes
linked with the Hb D-Punjab [β121(GH4) Glu→Gln, GAA>CAA] mutation in
eastern India, Hemoglobin. 2010;34(6): 530-7. https://doi.org/10.3109/01676830.2010.525900 PMid:21077760
- Adekile
A, Mullah-Ali, A. and Akar, N. A. (2010) Does elevated hemoglobin F
modulate the phenotype in Hb SD-Los Angeles?, Acta Haematologica,
123(3): 135-139. https://doi.org/10.1159/000276998 PMid:20110664
- Al-Barazanchi,
Z. A. A., Abdulateef, S. S., Hassan, M. K. and Jaber, R. Z. (2019)
Double heterozygosity for hemoglobin S and D Punjab in Basra, Iraq: A
Clinical and hematological study of 42 patients, Journal of Applied
Hematology, 10(4): 134-140. https://doi.org/10.4103/joah.joah_65_19
- Patel,
D. K., Purohit, P., Dehury, S., Das, P., Dutta, A., Meher, S., Patel,
S., Bag, S., Mashon, R. S. and Das, K. (2014) Fetal hemoglobin and
alpha thalassemia modulate the phenotypic expression of HbSD-Punjab,
International Journal of Laboratory Hematology, 36(4): 444-450. https://doi.org/10.1111/ijlh.12165 PMid:24245819
- Alkindi
S, Elsadek RA, Al-Madhani A, Al-Musalhi M, AlKindi SY, Al-Khadouri G,
Al Rawahi B, Al-Ruqeishi S, Al-Yazeedi J, Wali YA, Al Shamakhi S, Al
Rawahi M, Pathare AV.(2021) Impact of COVID-19 on vasooclusive crisis
in patients with sickle cell anaemia. Int J Infect Dis. 2021 Mar 16;
106 :128-133. https://doi.org/10.1016/j.ijid.2021.03.044 PMid:33741487 PMCid:PMC7962915
- Itano, H. A.: A third abnormal hemoglobin associated with hereditary hemolytic anemia. Proc. Nat. Acad. Sc. 37: 775-784, 1951. https://doi.org/10.1073/pnas.37.12.775 PMid:16589027 PMCid:PMC1063470
- Habara
A, Steinberg MH, Minireview: Genetic basis of heterogeneity and
severity in sickle cell disease, Exp Biol Med (Maywood). 2016 Apr;
241(7): 689-696. https://doi.org/10.1177/1535370216636726 PMid:26936084 PMCid:PMC4950383
- Alkindi
S, Al-Busaidi I, Al-Salami B, Raniga S, Pathare A, Ballas SK.,
Predictors of impending acute chest syndrome in patients with sickle
cell anaemia, Sci Rep. 2020 Feb 12;10(1):2470. https://doi.org/10.1038/s41598-020-59258-y PMid:32051480 PMCid:PMC7015921
- Alkindi
S, Al Zadjali S, Al Madhani A, Daar S, Al Haddabi H, Al Abri Q, Gravell
D, Berbar T, Pravin S, Pathare A, Krishnamoorthy R. Forecasting
hemoglobinopathy burden through neonatal screening in Omani neonates,
Hemoglobin. 2010; 34(2): 135-44. https://doi.org/10.3109/03630261003677213 PMid:20353348
- Fasola
F.A., Babalola O.A., Brown B.J., Odetunde A., Falusi A.G., Olopade O.
The effect of alpha thalassemia, HbF and HbC on haematological
parameters of sickle cell disease
patients in Ibadan, Nigeria.Mediterr J
Hematol Infect Dis 2022 , 14(1): e2022001, http://dx.doi.org/10.4084/MJHID.2022.001
- Daar
S, Hussain HM, Gravell D, Nagel RL, Krishnamoorthy R, Genetic
epidemiology of HbS in Oman: multicentric origin for the betaS gene Am
J Hematol, 2000, 64(1): 39-46. https://doi.org/10.1002/(SICI)1096-8652(200005)64:1<39::AID-AJH7>3.3.CO;2-R
- Al-Riyami AZ, Al-Marhoobi A, Al-Hosni S, Al Mahrooqi S, Schmidt M, O'Brien S, Al-Khabori M, Prevalence of Red Blood Cell Major Blood Group Antigens and Phenotypes among Omani Blood Donors, Oman Medical Journal [2019], Vol. 34, No. 6: 496-503. https://doi.org/10.5001/omj.2019.92 PMid:31745413 PMCid:PMC6851071