Pietro Tralongo*, Arianna Bakacs* and, Luigi Maria Larocca*.
Division of Anatomic
Pathology and Histology - Fondazione Policlinico Universitario
"Agostino Gemelli"- IRCCS, Università Cattolica del Sacro Cuore, Largo
Francesco Vito, 1, 00168, Rome, Italy.
*All authors contributed equally to this review.
Published: May 01, 2024
Received: February 29, 2024
Accepted: April 16, 2024
Mediterr J Hematol Infect Dis 2024, 16(1): e2024042 DOI
10.4084/MJHID.2024.042
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Epstein-Barr
virus (EBV) is a prevalent virus that can be detected in the vast
majority of the population. Most people are asymptomatic and remain
chronically infected throughout their lifetimes. However, in some
populations, EBV has been linked to a variety of B-cell
lymphoproliferative disorders (LPDs), such as Burkitt lymphoma, classic
Hodgkin lymphoma, and other LPDs. T-cell LPDs have been linked to EBV
in part of peripheral T-cell lymphomas, angioimmunoblastic T-cell
lymphomas, extranodal nasal natural killer/T-cell lymphomas, and other
uncommon histotypes. This article summarizes the current evidence for
EBV-associated LPDs in light of the upcoming World Health Organization
classification and the 2022 ICC classification.
|
Introduction
Epstein-Barr
virus-related lymphoproliferative diseases (EBV+ LPD) constitute a
diverse group of diseases characterized by the presence of the
Epstein–Barr virus (EBV) in one or more lymphoid cell types, resulting
in uncontrolled proliferation of infected cells. This phenomenon is
intricately linked to the development of a spectrum of
lymphoproliferative diseases (LPDs), ranging from non-cancerous to
precancerous and malignant conditions (Figure 1).[1-7]
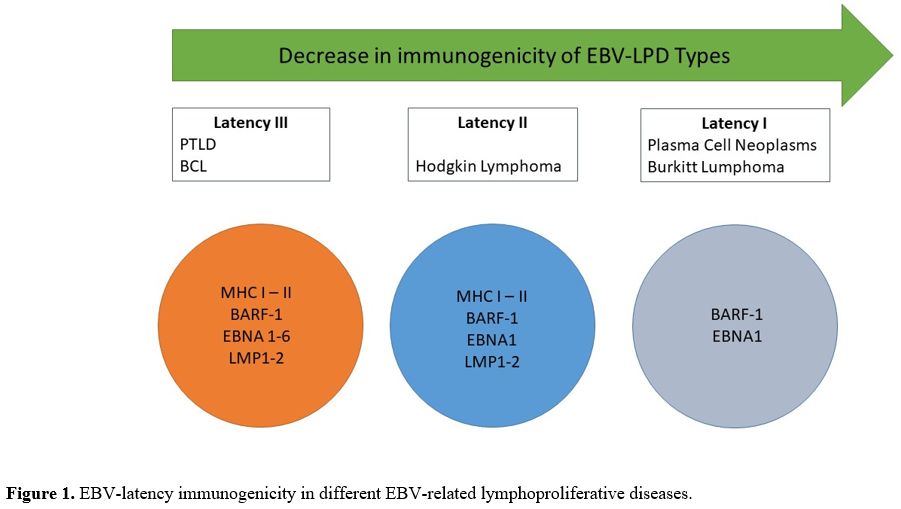 |
- Figure 1. EBV-latency immunogenicity in different EBV-related lymphoproliferative diseases.
|
Approximately 95% of the population is seropositive for EBV. Initial Infection with the virus may cause infectious
mononucleosis, present with minimal non-specific symptoms, or remain
entirely asymptomatic. Subsequently, the virus establishes latency
within its host, rendering the affected person asymptomatic for the
duration of their life. However, a small subset of
carriers - particularly those with immunodeficiency - develop an EBV+ LPD
weeks, months, years, or even decades after the primary Infection.
Globally, approximately 1% of all malignancies are attributed to EBV
infection, with LPDs constituting the great majority of these
malignancies.[1,8,9] Global health is greatly impacted by the non-malignant, premalignant, and malignant types of EBV+ LPD.[1]
This
review adheres to the categorization and nomenclature outlined by the
World Health Organization's 2016 modifications and the International
Collaboration on Cancer Reporting's 2022 refined criteria to elucidate
the complex landscape of EBV+ LPDs.[10-11] By
exploring these classifications comprehensively, we aim to enhance our
understanding of the diverse clinical entities within the realm of
EBV-associated LPD.
Pathophysiology
The
germinal center model describing the normal maturation of B cells
within lymph nodes and other lymphoid tissues delineates the
progression of naive B cells entering these structures into
lymphoblasts, centroblasts, centrocytes, memory B cells, and plasma
cells, ultimately acquiring the capability to produce functional
antibodies. Throughout this maturation process, B cells undergo
rearrangement of their immunoglobulin genes at various loci.[9] Notably, naïve B cells serve as the primary target for EBV invasion.
Upon
infiltrating naïve B cells, EBV orchestrates the expression of genes
that regulate cell maturation, compelling the cells to undergo forced
maturation, diminishing the infected cell's recognition by the host's
immune system, and triggering uncontrolled proliferation, culminating
in the development of a B cell-related LPD. The virus may also spread
from the initially infected B cells to T or NK cells, instigating
subsequent rounds of multiplication and the potential of LPD formation.[12]
Natural
killer T cells (NK), Gamma delta T cells, cytotoxic T cells (CTL),
helper T cells (Th), and follicular helper T cells (TFH) are
susceptible to EBV infection.[13] The mechanism by
which EBV infects dendritic-histiocytic cells (i.e. follicular
dendritic cells) adds further complexity to the understanding of the
viral dissemination. Although follicular dendritic cells are connective
tissue cells rather than lymphoid cells, their expression of the
surface membrane receptor CD21 serves as an entry point for EBV.
Through this CD21 entrance mechanism, EBV may potentially escape from
infected B cells and establish Infection within follicular dendritic
cells.[14]
It is crucial to emphasize that in
the majority of cases (>95%) EBV infections result in
non-complicated or asymptomatic outcomes. Host responses, particularly
involving T memory cells and CD8+ T cells, play a pivotal role in
controlling the outgrowth of infected B cells, confining the EBV genome
to episomes during the latency phase. However, in some cases, a
prevailing lytic phase can lead to a persisting EBV infection and a
chronic viral state influenced by diverse genetic and environmental
factors.
This influence is exemplified by the impact of ethnic and
geographical diversity on the occurrence of EBV-related malignancies.
Notably, specific Asian populations exhibit distinct genetic
predispositions, presumably linked to particular human leukocyte
antigen (HLA) types, influencing their response to primary EBV
infection. For instance, certain Asian ethnic groups with a heightened
prevalence of HLA A11, a type associated with an EBNA-4
mutation that hinders cytotoxic T-cell recognition of EBV, may
experience an aberrant response to the virus. In contrast, A11 is
uncommon among Europeans, and cytotoxic T cells recognizing the EBNA-4
peptide dominate the immunological response to EBV in Europeans. These
variations in the HLA phenotype offer a plausible explanation for the
higher prevalence of EBV-positive malignancies, particularly nasal T/NK
cell lymphomas, among Asian populations.[9]
Furthermore,
observations indicate that some immunocompetent hosts have the capacity
to eliminate EBV viral particles through oropharyngeal secretions
intermittently and asymptomatically over their lifetime. On the other
hand, immunodeficient hosts exhibit an increased frequency and quantity
of viral particles in oropharyngeal secretions, potentially leading to
increased EBV transmission.
EBV-Associated Reactive Lymphoid Proliferations
EBV-associated
reactive lymphoid proliferations represent a group of conditions
characterized by the growth of B cells or NK/T cells in response to EBV
infection. Typically, these conditions are self-limiting and
non-malignant, posing a low risk of progressing to malignant LPD.[1]
Epstein–Barr virus-positive infectious mononucleosis.
Approximately 90% of cases of infectious mononucleosis (IM) are
attributed to EBV. Beyond EBV, IM-like infections can result from human
cytomegalovirus, adenovirus, or other pathogens.[15]
acute EBV infection is frequently asymptomatic or mild in children
under the age of 5 years, while 25-75% of adolescents and adults
develop overt IM following Infection.[11] Within
weeks of EBV infection, signs and symptoms of IM appear. The majority
of cases show self-limiting flu-like symptoms such as fever, sore
throat, enlarged, painful lymph nodes in the head and neck, and
splenomegaly.
These symptoms normally resolve within six weeks. In
more severe cases, symptoms may persist longer and may be associated
with rare but serious complications such as hepatitis, anemia,
thrombocytopenia, hemophagocytosis, meningoencephalitis, myocarditis,
pericarditis, pneumonitis, parotitis, pancreatitis, and, in extremely
severe cases, life-threatening complications such as spleen rupture or
progression to other LPD such as hemophagocytic lymphohistiocytosis.[16-17]
During
the acute phase of EBV infection, individuals have high amounts of
infective EBV in their oral/nasal secretions, as well as high blood
levels of EBV, atypical lymphocytes, CD8+ T cells, and memory B cells
(up to 50% of the latter cells being EBV+). Tonsils and cervical lymph
nodes display hyperplasia, featuring a mix of normal-appearing
lymphocytes, activated lymphocytes, plasma cells, and
Reed-Sternberg-like cells.[15] Notably, many of these
normal-appearing and activated B cells, along with a small fraction of
the tissue's T and NK cells, are EBV+, with the virus predominantly
being in the lytic phase rather than latent.[1] Mild IM cases are often overlooked or diagnosed based on clinical and routine laboratory findings.
Diagnosis
of EBV-associated IM is conclusive when EBV, IgM antibody to EBV
viral-capsid antigen (VCA-IgM), IgG antibody to VCA (IgG-VCA), and IgG
antibody to EBV viral-capsid antigen (EBNA1-IgG) are detected in the
blood during the initial period of Infection and/or EBV is detected in
the oral or nasal secretions.[11] There are no
randomized controlled trials for the treatment of uncomplicated EBV+
IM. Patients experiencing airway obstruction, autoimmune reactions
(e.g., autoimmune anemia or thrombocytopenia), or other disease
consequences are commonly prescribed short-term corticosteroid regimes.[17]
Treatment of severe IM cases often entails regimens that are tailored
to the unique characteristics of each type of complication.[11]
Epstein–Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH).
EBV-HLH presents as a rare condition characterized by a systemic
inflammatory response or, in severe cases, as an overwhelming cytokine
storm. The virus impairs the capacity of cytotoxic T cells to eliminate
other EBV-infected cells, leading to excessive production of
pro-inflammatory cytokines, such as TNF-alpha, IL1beta, and CXCL9, by
activated histiocytes.[1]
Two forms of HLH
exist. Primary HLH (also known as genetic or familial HLH) is caused by
loss of function (i.e. inactivating) mutations in genes crucial for
cytotoxic T and/or NK cells to eliminate EBV-infected cells. These
mutations encompass UNC13D, STX11, RAB27A, STXBP2, LYST, PFP, SH2D1A, BIRC4, ITK1, CD27, and MAGT1 genes.[18]
Secondary
HLH is associated with and potentially aggravated by malignant and
non-malignant illnesses that, like primary HLH, impair the immune
system's ability to target EBV-infected cells. Hematological
malignancies, autoimmune disorders,[18] immunodeficiency disorders,[19-21] and infections are linked to secondary HLH.
The
typical manifestations of HLH include fever, decreased circulating
white blood cells and/or platelets, enlarged liver and/or spleen,
clinical signs of hepatitis, and/or central nervous system disorders[21]
such as irritability, decreased levels of consciousness, seizures,
meningitis, impaired cranial nerve function, hemiplegia, and ataxia.[18]
Histological
examination reveals infiltration of small EBV+ T cells, scattered EBV+
B cells, reactive histiocytes, reactive macrophages, and, in
approximately 70% of cases, hemophagocytosis-the ingestion of
erythrocytes, leukocytes, platelets, and/or their precursor cells by
histiocytes and macrophages-in various tissues such including
lymphatic, bone marrow, liver, and neuronal tissues. It is essential to
note that the presence of hemophagocytosis does not necessarily mean
that HLH has been diagnosed. Rather than being in a latent phase, the
EBV-infected lymphocytes are in their lytic cycle.[1]
The
combination of etoposide and dexamethasone is currently recommended for
the treatment of EBV+ HLH. Allogenic hematopoietic stem cell
transplantation is selectively employed after induction therapy,
especially in cases with primary HLH.[21] However, the success rate in managing EBV+ HLH remains lower than that of other secondary HLH causes.[11]
The use of anti-thymocyte globulin, the DEP regimen (liposomal
doxorubicin, etoposide, methylprednisolone), an anti-interferon gamma
monoclonal antibody, and especially rituximab are explored as novel
approaches to HLH, especially in cases of refractory or recurrent
disease.[11,23]
EBV-Positive T and NK Cell Lymphoproliferative Disorders of Childhood
Childhood
EBV+T and NK LPDs are a rare group of disorders that primarily affect
children, with sporadic occurrence in adults. The 2022 ICC has
implemented substantial revisions to this category of diseases.[11]
The prior 2017 WHO classification identified two categories: systemic
EBV+ T cell lymphoma of childhood and chronic active EBV (CAEBV)
infection with both cutaneous and systemic forms.[10]
The ICC 2022 now recognizes four distinct disorders: severe mosquito
bite allergy, hydroa vacciniforme (HV) LPD, systemic EBV-positive T
cell lymphoma of childhood, and systemic chronic active EBV (CAEBV)
disease. These recognitions are a result of new findings and a deeper
understanding of these disorders.[10]
Chronic active Epstein–Barr virus disease.
The term CAEBV disease is favored over CAEBV infection since most
adults harbor latent EBV infections displaying a persistent viral
presence, yet only a fraction develops the clinical condition.[11,23]
Characterized by a prolonged duration exceeding three months, CAEBV
disease progresses in the absence of known immunodeficiency,
manifesting as significantly elevated blood levels of EBV DNA and organ
involvement by EBV-infected cells.
About half of CAEBV patients
exhibit symptoms similar to infectious mononucleosis, such as
lymphadenopathy, hepatosplenomegaly, and fever.[24]
Additional symptoms include severe mosquito bite allergy, a pronounced
rash, hepatitis/hepatic failure, diarrhea, uveitis, myocarditis, and
interstitial pneumonia.[25] While the clinical course
varies, it tends to be protracted, with some patients exhibiting
prolonged stability., Notably, patients with EBV-infected T cells have
worse survival rates, more pronounced systemic symptoms, and elevated
blood EBV DNA titers compared to those with NK cell involvement.
Conversely, severe allergies to mosquito bites and elevated blood IgE
levels are common in patients with NK cell-related cases. In 2022, the
ICC designated these cases as CAEBV disease due to their aggressive
clinical course and absence of typical hydroa vacciniforme (HV)
lesions.[11,26] Currently, hematopoietic stem cell transplantation remains the sole curative treatment. Histopathological examination[27]
reveals infiltrating cells in affected organs without malignant
lymphoproliferation. A biopsy of the liver or lymph nodes is usually
conducted for diagnosis. Liver biopsy typically shows portal and
sinusoidal inflammation, indicative of viral hepatitis, while lymph
nodes may exhibit small granulomas, localized necrosis, or follicular
or paracortical hyperplasia. EBV infections affect 40% of NK cells and
60% of T cells. The majority of the invading T cells are CD4+, with a
small percentage being CD8+. Epstein-Barr virus-encoded small RNA
(EBER) is generally positive.[24] While the pathophysiology remains uncertain, genetic predisposition may play a role.[28]
TCR gene rearrangement may be monoclonal, oligoclonal, or polyclonal.
Recent genetic studies have identified somatic mutations in DDX3D and
KMT2D, particularly in cases of NK cell involvement, suggesting a
premalignant nature of the disease. Frequent intragenic deletions in
the EBV genome highlight the pivotal role of these mutations in the
pathogenesis of the disease.[29]
Severe mosquito bite allergy.
Severe mosquito bite allergy (SMBA) is a rare condition that primarily
affects young East Asians with a median age of onset at 6.7 years. In
most cases, SMBA is a symptom of CAEBV disease of the EBV+ NK cell
type, with approximately 33% of all CAEBV patients developing this
allergic response. SMBA has also been reported in rare cases of other
EBV EBV-related conditions such as Hodgkin lymphoma, hydroa
vacciniforme, aggressive NK cell lymphoma/leukemia), and extranodal
NK/T-cell lymphoma, nasal type. Additionally, it has been observed in
EBV-negative LPD, such as chronic lymphocytic leukemia and mantle cell
lymphoma.[30]
In the context of CAEBV, SMBA is
characterized by skin redness, swelling, ulceration, necrosis, and/or
scarring at the site of the bite. Concurrently, patients may experience
fever and malaise, enlarged lymph nodes, liver, and/or spleen, liver
dysfunction, hematuria, and proteinuria.[30] Severe cases may involve hemophagocytosis, NK/T-cell lymphoma, or aggressive NK cell leukemia.[31]
Pathologically, the skin lesions exhibit invading NK cells in the
epidermis and subcutaneous tissue, with a minor fraction of these cells
being EBV+, indicating that the virus is in its latency II phase. A
high density of EBV+ NK cells in these lesions may suggest that the
disease has progressed to NK/T cell lymphoma or NK cell leukemia.[1]
The cause of SMBA is not fully understood, but it is suspected that
mosquito salivary gland allergenic proteins cause EBV reactivation in
latently infected NK cells. This reactivation, facilitated by EBV
genes, such as LMP1, can lead to immortalization, proliferation, and, in rare cases, malignancy of EBV-reactivated NK cells.[30]
The optimum treatment for SMBA remains uncertain. Mild and
uncomplicated cases can be managed conservatively. In contrast, cases
showing evidence of substantial CAEBV disease, such as
hemophagocytosis, NK/T cell lymphoma, or aggressive NK cell lymphoma,
may require chemotherapeutic regimens.[30] Cases of
EBV+ SMBA with concurrent aggressive CAEBV have been treated with
success using a three-step CAEBV treatment regimen (immunotherapy,
cytoreduction, reconstruction).[32] Rare cases of
SMBA have been observed in individuals initially lacking an obvious
underlying condition but later developing CAEBV, highlighting the need
for thorough evaluation and follow-up in such cases.[30,33]
Hydroa vacciniforme-like lymphoproliferative disease.
Hydroa vacciniforme presents as an uncommon photodermatitis reaction
characterized by pruritic skin papules and vesicles that progress to
crusting and scarring upon exposure to sunshine—predominantly appearing
on sun-exposed facial and dorsal hand areas. This EBV+ condition
primarily affects children, demonstrating a variable course with
remissions and exacerbations before resolving in early adulthood.
However, adults can also be susceptible to this illness. In some
instances, the condition can progress to create severe, extensive, and
disfiguring skin lesions that are unrelated to sun exposure. Additional
manifestations include facial edema and systemic symptoms, such as
fever, weight loss, lymphadenopathy hepatomegaly, and/or splenomegaly.
Notably, EBV+ LPD, including T cell lymphoma, T cell leukemia, B cell
lymphoma, or B cell leukemia may arise.[34] The
milder and more aggressive forms of hydroa vacciniforme were initially
categorized as classic hydroa vacciniforme and hydroa vacciniforme-like
lymphoma, respectively. The extensive overlap between the two disease
types prompted the World Health Organization in 2016 to reclassify them
as Hydroa vacciniforme-like LPD, falling under the umbrella of CAEBV.
Histological examination of skin lesions reveals infiltrating
lymphocytes, predominantly T cells with a minority of NK- or B- cells.[34] EBV is commonly detected in T cells and, to a lesser extent, in NK cells of skin lesions.[31] Marker assays show that EBV is in latency phase II in these cells.[10]
Non-aggressive
cases of hydroa vacciniforme-like LPD typically respond to well to
conventional dermatological approaches suitable for non-malignant
diseases. Immunotherapeutic drugs, such as prednisone, interferon,
chloroquine, and thalidomide, can provide temporary remissions and
improvements in malignant cases of the disease. Standard chemotherapy
and radiotherapy protocols, commonly effective for lymphoma and
leukemia have shown only transient benefits in this context often
accompanied by severe toxicities.[34] The three-step
CAEBV treatment regimen has been employed with reasonable efficacy in
cases of EBV+ hydroa vacciniforme-like LPD where there is clear
evidence of concomitant CAEBV.[35]
Systemic Epstein–Barr virus-positive T cell lymphoma of childhood.
Systemic EBV-positive T cell lymphoma of childhood (TCLC) is a rare and
severe T cell lymphoma predominantly affecting children, adolescents,
and young adults, with a higher prevalence in Asian and Latin American
populations. The disease typically arises as a complication or
progression of either EBV-positive infectious mononucleosis (EBV+ IM)
or CAEBV disease.[1] The manifestations of TCLC occur
as a deterioration of signs and symptoms, either three weeks after the
onset of an EBV+ IM-like disease or at any point during CAEBV.
Clinically, TCLC presents with progressive hepatosplenomegaly,
worsening liver dysfunction, new skin rashes, pancytopenia,
hemophagocytosis in the bone marrow and spleen, coagulopathy, sepsis,
and potential organ failures. Notably, patients with TCLC show low or
undetectable levels of circulating IgM antibody but measurable amounts
of IgG antibody targeting EBV capsular antigens, distinguishing it from
the immunological profile seen in IM. Rapid proliferation of small or,
less commonly, large lymphoid cells in affected organs characterizes
the disease. These cells are EBV-positive cytotoxic T cells that
express CD8, CD3, CD2, TIA1, and granzyme, but not CD56. In the context
of CAEBV disease, these cells are usually CD4+ T cells or a mix of CD4+
and CD8+ T cells. The condition is usually lethal within a few weeks
after diagnosis. However, a subset of patients responded to the
HLH-2004 chemotherapy program involving agents such as etoposide,
dexamethasone, cyclosporine A, or, in certain cases, corticosteroids
and intrathecal methotrexate, with or without subsequent hematopoietic
stem cell transplantation.[31]
EBV+ NK/T Cell Lymphoproliferative Diseases
While
EBV primarily targets B cells, it can infect various immune cells,
including CD4+ T cells (T helper cells), CD8+ cells (cytotoxic T
cells), and natural killer (NK) cells, The mechanism by which EBV
spreads to these other cell types remains unclear, but it is postulated
that direct contact with virus-infected B cells facilitates such
infections.[1]
Extranodal NK/T cell lymphoma,
nasal type, peripheral T cell lymphoma, not otherwise specified (PTL,
NOS), and angioimmunoblastic T-cell lymphoma (AITL) are among the
NK-cell or T-cell malignancies that fall under the category of
peripheral T cell lymphomas (PTCL).
Extranodal NK/T cell lymphoma, nasal type.
Extranodal NK/T cell lymphoma, nasal type (ENKTL), is a type of NK or,
less commonly, T cell lymphoma that typically affects individuals of
Asian descent and indigenous populations in Mexico, Central America,
and South America. This malignancy typically manifests as tumors in the
nasal cavities, paranasal sinuses, palate, tonsils, nasopharynx,
hypopharynx, and/or larynx, with approximately 20% of cases exhibiting
tumors in the skin, soft tissues, gastrointestinal (GI) tract, testes,
and/or central nervous system (CNS). Affected individuals are typically
middle-aged and present with noticeable tumors, hemoptysis, ulcerating
skin nodules, upper airway obstruction, and/or obstructions/bleeding in
the lower GI tract, particularly in the colon. Lymph node involvement
is atypical and usually arises from tumor dissemination from their
initial locations.[1] Approximately 70% of ENLTL cases
are diagnosed with stage I or II diseases, indicating tumors that are
limited to a specific site or region of the body, with the remaining
cases having widespread stage III or IV disease.[36]
ENKTL is characterized by destructive, ulcerating, and necrotic lesions
at all stages. Histologically, the tumors comprise small, medium-sized,
or large malignant lymphoid cells, which are frequently accompanied by
a mix of benign inflammatory cells. Malignant cells express NK and/or T
cell markers (e.g., CD2, CD56, CD38), granzyme B, perforin, TIA1, and,
in the case of T cells, T-cell receptor gamma and delta chains.[31] TP53 and/or PD-L1 are often overexpressed.[13] In nearly all instances, the lymphoma cells are EBER+ and have a latency II pattern of EBV infection (Figure 2).[1]
 |
- Figure 2. Extranodal
NK/T cell lymphoma, nasal-type showing a diffuse infiltrate of lymphoid
cell of various sizes with angiotropism, destruction of vessels, and
necrosis (A, H/E, 100x). The neoplastic cells are diffusely and
strongly positive for EBER (B, EBER cISH, 100x).
|
They
harbor numerous somatic gene mutations, including commonly mutated
genes such as beta-catenin and ECSIT, involved in cell development and
survival.[31] Other genes, such as JAK3, STAT3, and STAT5B, associated with pro-malignant pathways, are altered in significantly fewer cases. Furthermore, epigenetic regulators (BCOR, KMT2D, ARID1A, EP300), tumor suppressor genes (TP53, MGA) and RNA helicase DDX3X are implicated.[37-40] A comprehensive investigation has identified seven genetic clusters associated with various clinical outcomes.[41] However, the relationship between EBV infection, these gene modifications, and the development of ENKTL remains unknown.[31]
Epstein–Barr virus–related peripheral T cell lymphoma, not otherwise specified.
Peripheral T cell lymphoma, not otherwise specified (PTCL, NOS), is an
unfavourable, heterogeneous category of T cell neoplasia defined by
characteristics that do not align with the diagnostic criteria for
other forms of PTCL.[10] PTCL, NOS accounts for
30-40% of all PTCL cases. This lymphoma most typically affects men with
a median age of 60 years - the majority of cases present in advanced
stage III or IV disease (70%). T-cell infiltration leads to widespread
lymph node swelling and concurrent bone marrow, liver, spleen, and/or
skin involvement.[42] Patients frequently exhibit B symptoms, including fever, night sweats, and weight loss.[43] Histologically, the involved tissues reveal mature T cells expressing CD4.[44]
Despite efforts to establish diagnostic criteria for PTCL and NOS based
on histology and immunophenotyping, these criteria have not been widely
implemented in clinical practice.[45] Various fusion rearrangements of VAV1 or TBX21 genes, as well as fusion rearrangements of the ITK gene with the SYK, FER, or ERBB4
genes, have been associated with PTLC and NOS. Two distinct profiles of
gene expression have been identified: malignant cells may overexpress
genes such as GATA3, MYC, mTOR, and β-catenin genes, or TBX21, interferon-, and NF-B genes. Individuals with GATA3 gene expression in their malignant cells have a lower overall five-year survival rate compared to those with TBX2 gene expression.[42]
In approximately 30% of PTCL, NOS cases are infected with EBV, with the
virus being in its latency II phase. Significant EBER expression in
malignant T cells is observed in only a few of these cases. EBER
expression is typically restricted to the small and large benign B
cells within the background of the disease's lesions. Consequently the
role of EBV in the initiation and progression of PTCL, NOS remains
unclear.[1]
Angioimmunoblastic T-cell lymphoma.
Angioimmunoblastic T cell lymphoma (AITL) is a systemic lymphoma
characterized by the presence of mature follicular helper T cells (TFH
cells).[1] Two other nodal T-cell lymphomas,
follicular T-cell lymphoma (FTCL) and nodal peripheral T-cell lymphoma
with the TFH cell phenotype (PTCL-TFH) have been described, both having
different morphologic features than AITL but sharing a TFH cell
immunophenotype as well as genetic and molecular characteristics.
Additionally, cutaneous T-cell proliferations with the TFH cell
phenotype have been reported.
AITL typically presents shortly
after antibiotic use, Infection, or allergic reactions. Common
manifestations include generalized lymph node swelling, enlarged liver
and spleen, skin lesions such as rash or, less usually, nodules,
plaques, purpura, and urticaria, bone marrow involvement, and B
symptoms such as fever, weight loss, and night sweats. Additional
manifestations may encompass arthralgias, arthritis, pleural effusions,
ascites, lung lesions, as well as neurological and gastrointestinal
symptoms. Common laboratory findings include immune-mediated hemolytic
anemia, elevated blood levels of eosinophils, gamma globulins, and LDH,
a high erythrocyte sedimentation rate (ESR) and positive blood tests
for autoantibodies like rheumatoid factor, anti-nuclear antibody, and
anti-smooth muscle antibody, suggesting an underlying immune system
problem. Histologically, involved tissues exhibit small lymphoid cells
surrounding venules against a background of TFH cells, activated
lymphocytes, follicular dendritic cells, epithelioid cells, plasma
cells, and eosinophils. Among these, only the TFH cells are malignant,
constituting 5-30% of all cells in the lesions. These malignant TFH
cells express specific markers, such as CD3, CD4, CD10, PD-1, and also
the B lymphocyte chemoattractant, CXCL13.[46] The
recent identification of TET2 and DNMT3A mutations in TFH lymphomas
motivated research into the link between two seemingly unrelated types
of hematologic malignancies. Interestingly, investigations have
revealed that TFH lymphomas can originate from divergent clonal
evolution from TET2 and DNMT3A-mutant progenitor cells after acquiring
RHO and/or IDH2 mutations. Notably, almost all cases show a dispersion
of EBV+ B cells, indicating that the virus is possibly in a confined
latency II phase. However, EBV is absent in the aggressive TFH cells
themselves. The EBV+ B cells carry non-malignant alterations, exhibit
excessive proliferation, and can potentially transform into EBV+ B cell
lymphomas.[1] Despite the implication of EBV in the
development and transition of these B cells, the precise role of the
virus in AITL and its association with the aggressive TFH cells remain
unknown.
Follicular T cell lymphoma.
The World Health Organization (2016) reclassified follicular T cell
lymphoma (FTCL), previously considered a variant of peripheral T cell
lymphomas, within the category of angioimmunoblastic T cell lymphoma
(AITL) and other nodal TFH cell lymphomas. This rare disorder shares
similarities with AITL as a lymph node-based malignancy or TFH cells.
However, it differs from AITL by allowing a diagnosis at an early,
limited, and comparatively less aggressive stage, with tissue lesions
lacking AITL-specific features, such as vascular proliferation.[1]
FTCL is predominantly observed in the elderly; however, it has been
documented in individuals as young as 27 years old. Common clinical
presentations involve advanced stage III or IV disease, characterized
by lymphadenopathy, hepato- and splenomegaly, and malignant cell
infiltrations in the bone marrow or, less commonly, tonsils, salivary
glands, and/or hard palate. B symptoms occur in a third of cases. A
positive Coombs test with or without autoimmune hemolytic anemia, high
blood LDH, and high gamma globulins are examples of laboratory
abnormalities.[47] Histopathologically, a follicular
lymphoma-like pattern is observed, in which malignant TFH cells form
nodules, and that is interpreted as a progressive transformation of a
germinal centre-like pattern, in which malignant TFH cells form
irregularly-shaped nodules surrounded by IgD-positive mantle cells.
These lesions may also contain large immunoblasts and, on rare
occasions, Reed-Sternberg cell-like B cells. In 50-60% of FTCL, one or
more of these B cell types, but not the malignant TFH cells, are
infected with EBV.[1] FTCL diagnosis relies on
clinical and laboratory findings, histopathology, and the presence of
TFH cells in lymph nodes, skin, or other lesions confirmed by
expression of relevant markers like PD-1, ICOS, CXCL13, CXCR5, and TOX.
Epstein–Barr virus-associated aggressive NK cell leukemia.
EBV-associated aggressive NK cell leukemia (EBV+ ANKL) is a rare NK
cell cancer that primarily affects Asians and young to middle-aged
individuals. It can also arise directly from other NK cell
proliferative diseases, such as chronic active EBV infection (CAEBV),
which is more common among the younger population.[1]
A study in China observed that almost all patients had B symptoms
(weight loss, fever, night sweats) and an enlarged liver and/or spleen
but no lymph node involvement. Laboratory studies revealed pancytopenia
in almost all cases, with small increases in the levels of circulating
large granular lymphocytes suspected to be malignant NK cells in 50% of
cases. Increased numbers of NK cells were found in the bone marrow in
all cases, accompanied by highly elevated blood levels of lactic acid
dehydrogenase and beta2 microglobulin. Liver damage was evident with
increased blood levels of enzymes, total bilirubin, and indirect total
bilirubin. All cases showed the presence of EBV+ cells in bone marrow
and tissue infiltrates, with circulating EBV+ lymphocytes in a few
cases.[48] Other studies reported EBV+ NK cells in 85-100% of patients.[1]
Histological examination of affected tissues revealed infiltrates of
large granular EBV+ NK cells mixed with benign inflammatory cells often
localized around small vessels, accompanied by tissue necrosis. With
EBV in latency II, the EBV+ NK cells express the CD56 antigen and are
malignant.[49] The LMP1 viral protein is expressed at
relatively high levels in NK cells, potentially activating the NF-kB
cell signaling system and stimulating the proliferation of EBV-infected
cells.[1] These findings are identified in 84% of
individuals who have "classic ANKL". "Sub-acute ANKL", affecting 16% of
individuals, manifests symptoms resembling infectious mononucleosis for
3-15 months before progressing to the aggressive course indicative of
classic ANKL.[50]
Intravascular NK/T-cell lymphomas.
Intravascular NK-cell lymphoma and intravascular T-cell lymphoma
represent two exceedingly rare forms of intravascular lymphomas caused
by EBV infection of NK- and cytotoxic T-cells, respectively. Afflicting
primarily young individuals, these conditions manifest with skin
lesions and signs of CNS involvement. In a minority of cases, there is
additional involvement in bone marrow, liver, kidneys, ovaries, and/or
cervix.[51] Affected individuals display signs and
symptoms of disseminated disease, such as fever, weight loss, night
sweats, arthralgias, jaundice, cytopenias, and multiple organ
involvement.[52] In general, both intravascular
lymphomas exhibit aggressive and rapid progression, with patients
typically responding poorly to treatment and having short life spans.[53-56]
Immunodeficiency - Associated Lymphoproliferative Disorders
Posttransplant
LPD (PTLD) and distinct LPD emerging in patients receiving
immunosuppressive therapy, including those associated with several
chemotherapeutic treatments, are examples of iatrogenic
immunodeficiency-associated LPD. They are included in this review
because a significant subset of them is EBV+; PTLDs were identified
more than 50 years ago and are recognized separately from lymphomas and
other iatrogenic immunodeficiency - related lymphoproliferative
disorders in the 2001, 2008, and 2016/2017 WHO classifications, as well
as in the 2022 ICC.[57-59] The 2022 ICC recommends a
similar classification for the latter entities. These LPDs, like PTLDs,
can be EBV+ or EBV-negative. It remains uncertain whether current
clinical guidelines for treating PTLD are applicable to patients with
other iatrogenic LPDs,[58-59] and some general
recommendations may be inappropriate in a non-transplant setting. This
holds true for biologic studies of PTLD, including investigations into
molecular or tumor microenvironmental characteristics.[60-62]
In
the context of identifying lymphoid proliferations in patients
following solid organ or stem cell transplantation, the 2022 ICC
recommends as a first step to determine whether the condition
represents a PTLD or whether another explanation exists, such as a
specific infection, a non-specific process, or, in the cases involving
the transplanted organ, whether it reflects rejection. In some
instances, both rejection and a PTLD may coexist. While a majority of
PTLD are EBV+, 20-40% are not, and there is an increasing number of
late EBV-negative PTLD g.[58] EBV+ patients typically
exhibit latency pattern III or, less commonly, pattern II. Notably, in
immunocompetent hosts, the presence of a limited number of EBV+ cells
do not necessarily imply the PTLD development, as scattered EBV+ cells
can be found in various lymphoid proliferations. After Following PTLD
confirmation, the next priority is subclassification. Excisional biopsy
is recommended for diagnosis, and re-biopsy of recurrent lesions is
advisable when feasible to rule out evolution or alternative causes
beyond PTLD.[58-59]
Non-destructive PTLDs.
Non-destructive PTLDs have a preserved underlying architecture
characterized by a small lymphocytic proliferation along with
hyperplastic and polytypic plasma cells. This includes
lymphoplasmacytic proliferation with prominent immunoblasts reminiscent
of IM in an immune-competent host, or the presence of a marked
follicular hyperplasia. As these histological patterns are non-specific
reactive proliferations, their detection is often reliant on
substantial EBV positivity, best visualized using EBER in situ
hybridization, or, less commonly, due to their conspicuous mass-forming
nature. Some individuals manifesting non-destructive PTLD may develop
overt PTLD at other sites either synchronously or metachronously.
Notably, non-destructive PTLD can harbor clonal populations with
cytogenetic and mutational abnormalities, although the majority do not
display such characteristics.[60-61,63]
Polymorphic PTLD.
Polymorphic PTLD represents the most prevalent yet challenging subtype
of PTLD, characterized by the architectural effacement of underlying
tissues. it is distinguished by the presence of variably sized and
shaped lymphoid cells, plasma cells, and immunoblasts some of which may
bear resemblance to Reed-Sternberg cells. In specific locations,
angioinvasion and necrosis may be observed. Notably, polymorphic PTLDs
are not expected to meet the criteria for lymphoma in an
immunocompetent host. These entities typically harbor clonal B cell
populations, with the clone size often being modest, and may exhibit
mutations, albeit not as frequently as observed in monomorphic PTLDs.[64]
The literature remains divided on whether patients with polymorphic
PTLD experience a more favorable prognosis compared to those with
monomorphic PTLD.[65]
Monomorphic PTLDs.
Monomorphic PTLDs bear resemblance to various lymphomas observed in
immune-competent hosts; however, not all cases are composed solely of
sheets of large cells, challenging the conventional understanding of
the term “monomorphic”. Presently, the term encompasses cases, that may
involve proliferations of monomorphic large cells or immunoblasts, with
some displaying a polymorphic appearance. B-cell monomorphic PTLD, for
instance, may exhibit a high concentration of reactive T cells or
plasmacytic differentiation, while other cases may consist primarily of
plasma cells. A substantial number of monomorphic T cell PTLD do not
manifest as sheets of large transformed cells. Furthermore, EBV+ MALT
lymphomas were included as a form of PTLD in the 2016–2017 WHO review (Figure 3).[66]
 |
- Figure 3. H&E
staining (A, 200x) of multiple fragments from a submandibular
lymph-node extensively replaced by a blastoid lymphoid population
consisting of medium to large sized elements with thick irregular
nuclei and prominent nucleoli, showing immunoreactivity for CD20 (B,
200x), CD5 (C, 200x), BCL-2 (D, 200x), cyclin D1 (E, 200x), SOX-11 (F,
200X) and concrete expression of EBER (G, 200x). Mitotic activity of
this post-transplant blastoid mantle cell lymphoma is dispersed with a
proliferative index (ki67) of 35-40% (H, 200x).
|
The
most prevalent monomorphic PTLDs resemble DLBCL, often falling it in
the non-germinal center B cell subtype; particularly in EBV+ patients.
Studies highlight both striking differences and similarities in
mutational and gene expression patterns between monomorphic PTLD of
DLBCL-type and related lymphomas in immune-competent hosts, in
particularly in EBV-negative patients.[64,67]
Noteworthy molecular and chromosomal changes seen in T/NKPTLD, as
opposed to B cell PTLDs, bear resemblance to those described for
peripheral T cell lymphomas in immunocompetent hosts.[68]
Burkitt lymphoma-type monomorphic B cell PTLD is a significant
diagnosis, as individuals with this condition may require immediate,
intensive therapy.[69-70] It is imperative to
distinguish monomorphic PTLD resembling plasma cell neoplasms from more
aggressive cases resembling multiple myeloma. Those resembling
plasmacytoma-like presentations may recover without the necessity for
intensive therapy.
Classic Hodgkin lymphoma PTLD.
Some PTLD are nearly always EBV+ and resemble classical Hodgkin
lymphoma (CHL), however they are uncommon in the posttransplant
scenario. Although it is advisable to proceed cautiously because many
other PTLD cases have cells that resemble Reed-Sternberg cells, this is
a specific diagnosis that must be made because it is a different type
of PTLD that is usually treated right away with conventional
CHL-therapy. Because the survival rates of monomorphic and polymorphic
PTLDs varies greatly, immunohistology tests are essential for ruling
out these conditions.[68] This is not the case for
other iatrogenic LPDs in patients with rheumatic disorders, where
Hodgkin-type LPD had a similar overall survival rate to DLBCL-type LPD
patients, but the PFS was worse in the Hodgkin cases.[71]
B-Cell Lymphoproliferative Diseases
Epstein–Barr virus-positive mucocutaneous ulcer.
EBV+ mucocutaneous ulcer is a rare lymphoproliferative condition
characterized by isolated, well-defined ulcers in mucous membranes and
skin caused by invading B lymphocytes.[1] Affected
individuals typically exhibit a compromised immune systems a due to
factors such as advanced age, immunosuppressive disorders (e.g.,
HIV/AIDS), immunosuppressive medication, or allogeneic hematopoietic
stem cell transplantation. Immunomodulatory drugs implicated in ulcer
formation include methotrexate, cyclosporin A, azathioprine,
mycophenolate, TNF inhibitors, tacrolimus, and topical steroids. The
diminished immune surveillance associated with certain predisposing
diseases or therapies is still sufficient to maintain systemic EBV
latency except where EBV+ B cells are prominent, such as in affected
mucous membranes and skin. Consequently, EBV+ cells in these locations
undergo unchecked multiplication, leading to tissue destruction and the
formation of ulcerative lesions.[72]
These
ulcers predominantly affect the elderly, typically presenting as
isolated lesions in the oral mucosa, and less frequently in the skin or
gastrointestinal tract mucosa. Individuals with EBV+ mucocutaneous
ulcer are usually asymptomatic and lack signs of lymphadenopathy or
involvement in other tissues, B symptoms, aside from localized pain at
the ulcer site and potential severe tissue degradation). However,
gastrointestinal ulcers can cause a variety of abdominal symptoms,
including perforations representing an acute emergency. Unlike most
other types of EBV+LPD, EBV-related mucocutaneous ulcers are not
associated with detectable EBV levels in the blood.[72]
Microscopically, these ulcers are composed of lymphocytes, including
EBV+ B cells and occasionally various other EBV+ lymphoid cell types.
In addition, histiocytes, plasma cells, eosinophils, and scattered
giant immunoblasts resembling, but distinct from the Reed-Sternberg
cells found in Hodgkin lymphoma are observed.[31]
These Reed-Sternberg-like cells are identified as EBV+ B cells
expressing the cell surface membrane protein, CD30, the B cell surface
membrane protein, CD20,[72] and EBV replication cycle latency II or III proteins.[1]
Epstein–Barr virus-positive Burkitt lymphoma.
Burkitt lymphoma is classified into three distinct types. Endemic
Burkitt lymphoma (eBL) is frequent in regions such as Africa, the
Middle East, Brazil, Papua New Guinea, and other malaria-endemic areas.
Typically, presenting in children aged 4 to 7 years, eBL is
consistently associated with EBV infection.[73]
Sporadic Burkitt lymphoma (sBL) is extremely uncommon, affecting
primarily children and, less frequently, adults over the age of 60.[31] with its main occurrence in Northern and Eastern Europe, East Asia, and North America.[74] In the United States, an estimated 1,200 cases are reported annually.[30] Only about 10-15% of sBL cases are linked to EBV infection.[75]
The immunodeficiency-related form of Burkitt lymphoma (iBL) afflicts
30-40% of individuals with HIV-induced AIDS31 and, in rare cases,
individuals who have undergone a bone marrow or organ transplant. In
the latter cases, individuals have consistently received
immunomodulatory drugs and are thus immunocompromised.[73] Approximately 30% of iBL cases are EBV-associated.[76]
A
jaw mass, periorbital swelling resulting from an orbital tumor, or an
abdominal mass attributed to a tumor in the retroperitoneum, kidney, or
ovary are all frequently observed in eBL. Additionally, eBL may present
with the less common manifestation of a sudden onset of paraplegia or
urine incontinence indicating tumor penetration into neural tissue. sBL
is characterized by abdominal discomfort, nausea, vomiting, and/or
gastrointestinal bleeding, all stemming from the growth of an abdominal
tumor, a head or neck tumor involving lymph nodes, tonsils, nose,
sinuses, and/or oropharynx or substantial bone marrow infiltration by
malignant tumor cells.[73] Fever, along with other
constitutional symptoms, and the presence of malignant disease in the
gastrointestinal tract, bone marrow, liver, lung, and central nervous
system are common indicators of iBL.[77] Histologic
examination of BL-involved tissues reveals infiltrations by a uniform
population of rapidly proliferating lymphocytes with a high mitotic
index approaching 100%, creating intermittent clear spaces resembling a
"starry sky" pattern due to macrophages containing ingested dead cells.
The predominant lymphocytes are B cells expressing CD20 and CD10
antigens, with a background of few T cells.[73] These
B cells predominantly derived from germinal center B cells, exhibit EBV
in latency I, and express substantial quantities of EBNA1 and EBER
viral proteins. Under certain circumstances, EBNA and LMP2A products are also expressed.[1]
The protein EBNA1 and EBER may contribute to the development and/or
progression of BL by blocking apoptosis in the infected cells, while
the product of LMP2A may stimulate the PI3K cell signaling pathway, boosting cell proliferation.
Malignant B cells in all three variants of BL frequently exhibit chromosomal translocations affecting the MYC gene. MYC
situated on the long arm of human chromosome 8 (8q24), is a
proto-oncogene that can induce cancer when properly mutated or
overexpressed. (). The translocations involve MYC
relocated to the IGH (immunoglobulin heavy chain) gene locus at 14q32,
the IGK (immunoglobulin kappa light chain) gene locus at 2p12, or the
IGL (immunoglobulin lambda light chain) gene locus at 22q11. These
translocations place MYC under the transcriptional control of these
antibody-forming loci, leading to the overexpression of the MYC
product, enabling the cells to undergo uncontrolled multiplication.
Concurrently, other genes within the BL cells may be undergo
alterations; for instance, approximately, 30% of BL cases exhibit
changes in the TP53 gene, which may improve cell survival.[31]
Due to these alternative pathways to malignancy, some of which may be
EBV-independent, and considering that not all BL cases involve EBV,
many cases of EBV+ BL are probably not solely caused or promoted by
EBV. While the ubiquitous virus is likely the cause of almost all cases
of eBL, it may act as an innocent passenger virus in numerous instances
of sBL and iBL.[1] EBV infection is usually associated
with increased mutation load, with type 1 EBV having a larger
mutational burden than type 2. Although sporadic and
immunodeficiency-associated BLs revealed comparable genetic profiles,
endemic BLs had more mutations in BCL7A and BCL6, but less in DNMT1, SNTB2, and CTCF. Silencing mutations in ID3 were seen in all three BL subtypes. In vitro mass spectrometry-based proteomics revealed that the ID3 protein predominantly binds to TCF3 and TCF4. In vivo deletion of ID3 enhanced the effects of MYC, resulting in fast carcinogenesis and tumor characteristics similar to those seen in human illness.
Epstein–Barr virus-positive lymphomatoid granulomatosis.
EBV+ lymphomatoid granulomatosis (EBV+ LG) is an uncommon condition
characterized by the coexistence of malignant B cells and reactive,
non-malignant T cells, almost invariably associated EBV+.[1]
This LPD primarily affects middle-aged males with a male-to-female
ratio of 2:1. Clinically, EBV+ LG typically presents as a lung
manifestation with coughing, hemoptysis, shortness of breath, and chest
X-rays revealing multiple nodular lesions at the base of both lungs
observe in approximately 90% of cases. Additionally, manifestations may
extend to nodular or infiltrative lesions in the skin, central nervous
system, kidney, liver,[1] and/or peripheral nervous system. Notably, lymph nodes are uninvolved at initial presentation[1] and in certain instances, lung involvement may be absent.[78] EBV+ LG lesions are characterized by the presence of sporadic large, atypical B cells[79]
amidst a background of abundant reactive CD4+ Helper T cells, plasma
cells, macrophages, and a variable number of giant atypical lymphoid
cells resembling immunoblasts, plasmablasts, or Reed-Sternberg cells.
The lesions frequently center on and exhibit damage of small blood
vessels but they lack well-formed granulomas.[78]
Within the lesions, only lymphoid B cells are positive for EBV,
expressing the viral proteins LMP1 and EBNA2 indicative of latency III
phase (Figure 4).[1]
 |
- Figure 4. Lymphomatoid
granulomatosis shows a polymorphous infiltrate of T-lymphocytes,
macrophages, and plasma cells that displays an angiocentric and
angiodestructive pattern, admixed with EBER+ Reed-Sternberg-like
B-lymphoid cells (A: H/E, 100x; B: EBER cISH, 100x).
|
Individuals afflicted by EBV+ LG may have compromised immune function due to subtle reductions in immune activity[1] or, as indicated by individual case reports, underlying immunodeficiency diseases.[78]
Case studies also suggest a potential association with inflammatory or
autoimmune disorders.[80] In skin manifestations of the disease,
non-malignant or malignant lymphoid proliferations may progress to or
be complicated by EBV+ LG.[81] The pathogenesis of
EBV+ LG involves in part the virus infecting B cells, which generate
chemokines that attract and activate T lymphocytes to cause tissue
damage, particularly affecting blood vessels. Impaired host immune
activity, coupled with the infected cells' failure to produce viral
proteins recognized by cytotoxic T cells, facilitates the evasion of
detection and proliferation of EBV+ B cells.[78]
LG
is stratified into three grades based on the histological
characteristics of biopsied tissues: grade I (<5 EBV+ cells per high
power microscopic field (hpf), no atypical cells/hpf, and minimal
necrosis); grade II (5-20 EBV+ cells/hpf, occasional atypical
cells/hpf, and moderate necrosis); and grade III (>20 EBV+
cells/hpf, predominance of atypical cells/hpf, and extensive necrosis).
Epstein–Barr virus-positive Hodgkin lymphoma.
Hodgkin lymphoma (HL) is classified into two histologic subtypes:
nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and classical
Hodgkin lymphoma (cHL), with cHL subtypes including nodular sclerosis
(NSHD), mixed cellularity (MCHL), lymphocyte rich (LRHL), and
lymphocyte depleted (LDHL). EBV is detected in 30% to 50% of cHL cases,
but only in 10% cases of LRHL, or NLPHL cases. The infiltrating cells
in HL encompass T cells, B cells, macrophages, eosinophils,
fibroblasts, surrounding the Reed-Sternberg cells (HRS cells). HRS
cells which are large mono- or multinuclear cells derived from lymph
node and/or spleen germinal center B cells are the sole malignant cells
in HL. In approximately 30-50% of HL cases, these cells may harbor EBV
and express viral products indicative of stage II latency (Figure 5).[40]
 |
- Figure 5. EBV-positive
Hodgkin lymphoma comprises a polymorphous infiltrate of T- and B-cells,
eosinophils, macrophages, and fibroblasts surrounding HRS cells (A:
H/E, 100x). Neoplastic cells are strongly EBV-positive (B: EBER cISH,
100x).
|
The role of EBV in the pathogenesis of EBV+ HL involves overexpression of the virus's LMP1 gene in HRS cells. LMP1 mimics activated human TNF receptors sustaining continuous stimulation of NF-kB, PI3K, and JAK-STAT
signaling pathways. This continuous activation promotes cell
proliferation, survival, and the production of cytokines that may
suppress the EBV's lytic cycle, thus maintaining the viability of HRS
cells.[40] HRS cells also express the virus's LMP2A gene product, which mimics the human BCR gene product, aiding in cell survival.[1]
Additionally, the presence of EBV in HRS cells associates with
crippling mutations in the rearranged immunoglobulin (Ig) genes,
inhibiting Ig expression and prompting HRS cells to secret cytokines
and chemokines. These mediators recruit various cell types into the
pathogenic infiltrates of EBV+HL, creating a local milieu that enables
HRS cells to evade the immune system and proliferate.[82]
EBV+
HL is more prevalent in children and young adults, but it can also
develop in individuals at older age, possibly due to age-related
decline in immune system function, infectious illnesses, or
malnutrition.[1] The incidence of EBV+ HLs in HIV/AIDS
patients is substantially higher, approximately 10-fold, compared to
the normal population, although the specific causes remain unknown.[40]
Symptoms of EBV+ HL are comparable to EBV-negative HL, including fever,
night sweats, weight loss in the presence of swollen lymph nodes, and
indications of tumor invasion into other organs.
Epstein–Barr virus-positive diffuse large B cell lymphoma, NOS.
Diffuse large B-cell lymphoma (DLBCL) stands as the most prevalent form
of lymphoma, primarily affecting elderly people, with comparatively
lower incidence in younger individuals and infrequent occurrences in
children. In addition to swollen lymph nodes, elderly individuals often
present with symptoms arising from malignant cell infiltrations into
the upper gastrointestinal tract, lungs, upper airways, and/or other
organs. In contrast, younger patients typically exhibit enlarged lymph
nodes but rarely have B symptoms or involvement of extranodal tissues.
The disease tends to be more aggressive in the elderly.[31]
Traditionally, DLBCL was classified based on the cell types in tissue
infiltrates into three patterns: the anaplastic variant (3% of cases)
characterized by Reed-Sternberg-like cells[83]
embedded in a background of histiocytes and lymphocytes; the
immunoblastic variant (8-10% of cases) dominated by 90% immunoblasts;
and the centroblastic variant (80% of cases) characterized by a
prevalence of centroblasts.[83] These histological
characteristics are usually accompanied by the invasion and destruction
of small blood vessels. The current classification is based on the cell
of origin of the disease, resulting in germinal center B cell DLBCL
(GCB-DLBCL) and activated B cell DLBCL (ABC-DLBCL). Recent
breakthroughs in genetic technologies, including next-generation
sequencing, have uncovered multiple recurrent genetic anomalies in
DLBCL. Two separate groups have presented large-scale genomic studies
with clinical data. These investigations developed new molecular
classifications based on various genetic anomalies associated with
DLBCL prognosis. A multi-omics investigation of over 300 DLBCL cases
was conducted, finding that DLBCL may be classified into five groups
(C1, C2, C3, C4, and C5) based on the combination of recurrent genetic
anomalies. This enabled risk classification for DLBCL patients and
promoted focused therapy. For example, C5 cases typically exhibit CD79B and MYD88-L265P mutations, ABC-DLBCL cases have poor outcomes, and sensitivity to BTK
inhibitors or lenalidomide has been found. DLBCL is less reliant on its
microenvironment, which is consistent with a total breakdown of normal
lymphoid structure. However, like with other B-cell lymphomas, recent
data suggests that the immune system is critical to the development and
outcome of DLBCL. In DLBCL, disturbed cross-talk between lymphoma cells
and the microenvironment contributes to lymphoma cells' ability to
evade immune monitoring by the host. Immune escape methods include
concealing from the immune system by deleting or lowering recognition
molecules (B2M, MHC-I, MHC-II), dampening antitumor immune activity (i.e. CREBBP, EZH2),
and generating a microenvironment that promotes lymphoma growth.
Altering immune recognition, in particular, plays a significant role in
DLBCL tumor formation and progression, and its molecular basis is being
studied extensively.[31] Uncommonly, DLBCL can arise
through a Richter transition of chronic lymphocytic leukemia (CLL) to
an exceedingly aggressive type of DLBCL. This transition is
particularly observed in EBV-associated CLL cases, constituting 10-15%
of all CLL cases. However, it should be noted that a significant number
of Richter syndromes is not EBV-related, since aberrant TP53 mutations are present with subclonal selection, in particular following chemoimmunotherapy.[84]
Patients with EBV-positivity account for around 10-15% of DLBCL cases.
EBV+ DLBCL, not otherwise defined (EBV+ DLBCL) is more common in East
Asia and Mexico. Distinguishing features of EBV+ DLBCL include the
expression of EBV genes typical for the virus's latency III (common in
the elderly) or II (common in younger individuals) by nearly all large
B cells.[75] These centroblastic B-cells express EBER,[31] LMP1, EBNA1, EBNA2, and other viral proteins[1]
along with traditional B cell antigenic markers such CD20, BCL6, and
CD19 in more than 50% of patients. The viral proteins are postulated to
activate signaling pathways such as NF-kB, STAT/JAK, NOD-like receptor, and TLR in infected cells, potentially enhancing cell proliferation and survival.[1]
EBV+
DLBCL is notably prevalent in immunocompromised individuals, and its
occurrence in the elderly is hypothesized to be linked to
immunosenescence. Immunosenescence includes an age-related decline in
specific types of CD4+ and CD8+ lymphocytes that function to suppress
EBV+ cell development.[1] Additionally, EBV+ DLBCL can arise in
immunocompromised individuals due to conditions such as HIV/AIDS, where
33% of patients are EBV+ or as a consequence of anti-rejection drug
therapy post-solid organ transplantation, where 30% to 70% of these
cases are EBV+.[83] Similarly, the Richter transition of EBV+ CLL to EBV+
DLBCL is observed in CLL cases treated with immunosuppressive
medications, indicating a correlation with immunosuppression-related
reactivation of latent EBV infection of these CLL cells.[84]
Epstein–Barr virus-associated diffuse large B cell lymphoma associated with chronic inflammation. DLBCL associated with chronic inflammation (DLBCL-CI) is an exceptionally rare subtype of EBV-positive DLBCL[1]
typically presenting as a tumor in areas characterized by chronic
inflammation, often occurring within bodily cavities or confined
spaces.[85] The majority of reported cases of
DLBCL-CI involve pyothorax-associated lymphoma (PAL). PAL develops
years after pneumothorax is intentionally induced for therapeutic
purposes, such as collapsing a lobe or complete lung around a cavity[85] or to manage pleurisy[86]
resulting from an uncontrollable underlying condition, most commonly
pulmonary tuberculosis. This distinctive lymphoma subtype has been
predominantly observed in older Japanese males. DLBCL-CI seldom occurs
in conjunction with other chronic inflammatory disorders such as
osteomyelitis, medical insertion of a foreign body (such as
intrauterine contraceptive devices, metallic implants, surgical mesh),
as well as skin ulcers and venous ulcers. The clinical manifestations
of DLBCL-CI represent the destructive consequences of the cancer in the
affected areas. Infiltrative lesions comprise a combination of benign,
EBV-negative chronic inflammatory white blood cells and diffuse large
EBV+ B cells in latency III. EBV+ large B cells in these lesions
frequently exhibit reduced expression of the CD20 antigen and genetic
abnormalities such as TP53 mutations, MYC overexpression, and TNFAIP3
deletion, at variance from alterations found in other EBV+ DLBCL NOS.
Research indicates that the condition is initiated by the EBV-driven
proliferation of large, activated B cells in a confined anatomical
area, thereby isolating them from immune surveillance.[15]
Additionally, the EBV-induced release of anti-inflammatory cytokines,
such as Interleukin 6 and Interleukin 10, may further facilitate
infected cells in evading the immune system.[1]
Fibrin-associated diffuse large B cell lymphoma.
The World Health Organization classified fibrin-associated diffuse
large B cell lymphoma (FA-DLBCL) as a form of DLBCL-CI in 2016. This
exceedingly rare disease exclusively affects immunologically competent
individuals.[1] FA-DLBCL is characterized by large B
cells infiltrating into long-standing, avascular fibrin-based masses
which develop in or around various structures, such as long-standing
hamartomas, pseudocysts, cardiac myxomas, prosthetic heart valves,[1] thrombus-laden endovascular grafts, hematomas,[31] hydroceles, and hip prosthetic implants.[87]
The infiltrations consist of sheets, bands, or clusters of
proliferating large B cells within avascular tissue covered with or
containing abundant fibrin. Notably, there is a scarcity or absence of
other types of inflammatory cells that comprise the infiltrations.[87]
Furthermore, rare B-cell lymphomas related to breast implants were
observed and were either classified as DLBCL-CI or FA-DLBCL. These
B-cell forms are often EBER+.[88] The large B cells are infected with EBV, which is present In latency III, expressing the virus's EBER, EBNA2, and LMP-1 genes.[31]
Importantly, these infiltrations rarely extend beyond the initial
sites, with no involvement of lymph nodes, spleen, or other tissues.
FA-DLBCL represents a benign expansion of EBV+ large B cells. Similar
to DLBCL-CI, FA-DLDCL localized immune suppression at the sites of
origin may contribute to the development of FA-DLBCL. However, in
contrast to DLBCL-CI, the large B cells in FA-DLBCL appear to be unable
to proliferate and survive long-term outside of the sequestered areas.
FA-DLBCL does not exhibit a highly malignant nature.[31] The large EBV+ B cells in FA-DLBCL, unlike those in DLBCL-CI, do not overexpress the MYC gene and have few karyotype chromosomal abnormalities.[87]
Patients
with FA-DLBCL present with signs and symptoms corresponding to the
location of the infiltrative lesion. Cardiovascular symptoms, such as
stroke, may occur when lesions affect the heart or vessels, such as
myxoma or prosthetic valves. Besides these cardiovascular consequences,
the disease typically progresses slowly and remains localized to the
site of origin. The curative approach often involves the surgical
removal of affected tissues and any associated foreign implant.
Epstein–Barr virus-positive plasmablastic lymphoma.
Plasmablastic lymphoma (PBL) is a rare lymphoma that predominantly
affects immunocompromised individuals, particularly those with
HIV/AIDS, marking it as an AIDS-defining clinical condition.[31]
Additionally, individuals who have undergone an organ transplant,
received chemotherapy, or are subject to age-related immunological
senescence are susceptible to PBL.[89] Chronic
autoimmune or inflammatory disorders, such as rheumatoid arthritis,
Graves' disease, giant-cell arteritis, sarcoidosis, or severe
psoriasis, have been implicated in the development of PBL.[90]
This condition can affect people of all ages, with a male-to-female
ratio of 4:1. PBL typically manifests as a tumor of the head and neck
region, gastrointestinal system, skin, or other tissues.
Histologically, PBL is classified into two types: monomorphic PBL,
primarily composed of immunoblastic cells, and plasmacytic PBL, which
consists primarily of plasma cells at various stages of development.
Despite their B cell origin, these cells display plasma cell markers
such as CD79a, IR4, BLIMP1, CD38, and CD138, while typically showing a
CD20 negativity.[31] Approximately 70% of PBL cases
are EBV-positive, with the majority of lymphoma cells expressing EBV
genes, indicating that the virus is in latency phase 0 or I.[1]
The disease appears to develop and progress as a result of the actions
of both EBV and the human immunodeficiency virus (i.e. HIV). PBL, in
particular the EBV-positive form, is associated with the overexpression
of the MYC gene, emphasizing
the role of the Myc protein in driving the disease. However, the
precise contribution of EBV in MYC gene overexpression, as well as its
role in the initiation and progression of EBV-positive PBL, remains
unclear.
Epstein–Barr virus-associated plasma cell myeloma.
Plasma cell myeloma (PCM) is malignancy characterized by the
infiltration of malignant plasma cells into the bone marrow or
development of soft tissue masses known as plasmacytomas. EBV may be
associated with this condition in rare cases, particularly in
individuals with immune deficiencies (e.g., HIV/AIDS, history of organ
donation) or chronic inflammation (e.g., rheumatoid arthritis).
Notably, EBV positivity is more prevalent in plasmacytoma than in PCM
with bone marrow infiltration.[1] Tissues affected by
EBV+ PCM often exhibit foci of EBV+ cells resembling immature or poorly
differentiated anaplastic plasma cells with a high proliferation rate.[1] The cells produce EBV gene products such as EBER,[56] indicating that EBV is in a confined latency II phase.[1]
Although these cells are generated from B cells, they exhibit plasma
cell markers rather than B cell markers. The specific role of EBV in
the development and evolution of EBV+ PCM remains uncertain.[15]
In comparison to individuals with EBV-negative illness, patients with
localized plasmacytoma and positive EBER are more likely to progress to
the infiltrative (i.e. systemic) type of PCM.[91]
Conclusion
The
new ICC and WHO classifications include numerous new entities and ideas
for EBV-positive LPDs, reflecting advances in genetics and molecular
virology. With a better understanding of LPD's clinical and pathologic
entities, we can perform EBV-ISH to detect clinically borderline
Infection and neoplastic conditions, a history of recurrent
inappropriate immune response, particularly in children, young adults
or the elderly, and a pathologically polymorphous inflammatory
background. The ultimate goal is to acquire accurate diagnosis and
management of EBV-associated conditions, with some entities requiring a
better understanding of histological and molecular features.
References
- Rezk SA, Zhao X, Weiss LM (June 2018). Epstein-Barr
virus-associated lymphoid proliferations, a 2018 update. Human
Pathology. 79: 18-41. doi:10.1016/j.humpath.2018.05.020. S2CID
47010934. https://doi.org/10.1016/j.humpath.2018.05.020
PMid:29885408
- Bjornevik K, Cortese M, Healy BC, Kuhle J,
Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, Ascherio
A (January 2022). Longitudinal analysis reveals high prevalence of
Epstein-Barr virus associated with multiple sclerosis. Science. 375
(6578): 296-301. Bibcode:2022Sci...375.296B.
doi:10.1126/science.abj8222. S2CID 245983763.
https://doi.org/10.1126/science.abj8222 PMid:35025605
- Ascherio
A, Munger KL (2015). EBV and Autoimmunity. Epstein Barr Virus Volume
1. Current Topics in Microbiology and Immunology. Vol. 390. pp. 365-85.
doi:10.1007/978-3-319-22822-8_15. ISBN 978-3-319-22821-1.
https://doi.org/10.1007/978-3-319-22822-8_15 PMid:26424654
- Naseem
M, Barzi A, Brezden-Masley C, Puccini A, Berger MD, Tokunaga R,
Battaglin F, Soni S, McSkane M, Zhang W, Lenz HJ (May 2018). Outlooks
on Epstein-Barr virus associated gastric cancer. Cancer Treatment
Reviews. 66: 15-22. doi:10.1016/j.ctrv.2018.03.006.
https://doi.org/10.1016/j.ctrv.2018.03.006 PMid:29631196
PMCid:PMC5964025
- Weiss RA (October 2016). Tumour-inducing viruses. British Journal of Hospital Medicine. 77
(10): 565-568. doi:10.12968/hmed.2016.77.10.565.
https://doi.org/10.12968/hmed.2016.77.10.565 PMid:27723397
- Mastria
G, Mancini V, Viganò A, Di Piero V (2016). Alice in Wonderland
Syndrome: A Clinical and Pathophysiological Review. BioMed Research
International. 2016: 8243145. doi:10.1155/2016/8243145.
https://doi.org/10.1155/2016/8243145 PMid:28116304 PMCid:PMC5223006
- Nussinovitch M, Prais D, Volovitz B, Shapiro R,
Amir J (September 2003). Post-infectious acute cerebellar ataxia in
children. Clinical Pediatrics. 42 (7): 581-4.
doi:10.1177/000992280304200702. S2CID 22942874.
https://doi.org/10.1177/000992280304200702 PMid:14552515
- Houldcroft
CJ, Kellam P (March 2015). Host genetics of Epstein-Barr virus
infection, latency and disease. Reviews in Medical Virology. 25 (2):
71-84. doi:10.1002/rmv.1816. https://doi.org/10.1002/rmv.1816
PMid:25430668 PMCid:PMC4407908
- Farrell PJ (August
2018). Epstein-Barr Virus and Cancer. Annual Review of Pathology. 14:
29-53. doi:10.1146/annurev-pathmechdis-012418-013023. S2CID 52051261.
https://doi.org/10.1146/annurev-pathmechdis-012418-013023 PMid:30125149
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein
H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES
(May 2016). The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127 (20): 2375-90.
doi:10.1182/blood-2016-01-643569.
https://doi.org/10.1182/blood-2016-01-643569 PMid:26980727
PMCid:PMC4874220
- Campo E, Jafe ES, Cook JR
et al (2022) The international consensus classification of mature
lymphoid neoplasms: a report from the clinical advisory committee.
Blood 140(11):1229-1253. https://doi.org/10.1182/blood.2022015851
PMid:35653592 PMCid:PMC9479027
- Worth AJ,
Houldcroft CJ, Booth C (November 2016). Severe Epstein-Barr virus
infection in primary immunodeficiency and the normal host. British
Journal of Haematology. 175 (4): 559-576. doi:10.1111/bjh.14339. S2CID
10779427. https://doi.org/10.1111/bjh.14339 PMid:27748521
- de
Mel S, Soon GS, Mok Y, Chung TH, Jeyasekharan AD, Chng WJ, Ng SB (June
2018). The Genomics and Molecular Biology of Natural Killer/T Cell
Lymphoma: Opportunities for Translation. International Journal of
Molecular Sciences. 19 (7): 1931. doi:10.3390/ijms19071931.
https://doi.org/10.3390/ijms19071931 PMid:29966370 PMCid:PMC6073933
- Dalia S, Shao H, Sagatys E, Cualing H, Sokol L
(October 2014). Dendritic cell and histiocytic neoplasms: biology,
diagnosis, and treatment. Cancer Control. 21 (4): 290-300.
doi:10.1177/107327481402100405.
https://doi.org/10.1177/107327481402100405 PMid:25310210
- Kunitomi
A, Hasegawa Y, Asano N, Kato S, Tokunaga T, Miyata Y, Iida H, Nagai H
(May 2018). EBV-positive Reactive Hyperplasia Progressed into
EBV-positive Diffuse Large B cell Lymphoma of the Elderly over a 6-year
Period. Internal Medicine (Tokyo, Japan). 57 (9): 1287-1290.
doi:10.2169/internalmedicine.9112-17.
https://doi.org/10.2169/internalmedicine.9112-17 PMid:29279478
PMCid:PMC5980812
- Mammas IN, Greenough A,
Theodoridou M, Kramvis A, Christaki I, Koutsaftiki C, Koutsaki M,
Portaliou DM, Kostagianni G, Panagopoulou P, Sourvinos G, Spandidos DA
(January 2016). Current views and advances on Paediatric Virology: An
update for paediatric trainees. Experimental and Therapeutic Medicine.
11 (1): 6-14. doi:10.3892/etm.2015.2890.
https://doi.org/10.3892/etm.2015.2890 PMid:26889211 PMCid:PMC4726865
- Dunmire SK, Verghese PS, Balfour HH (May 2018). Primary Epstein-Barr virus infection. Journal of Clinical Virology.
102: 84-92. doi:10.1016/j.jcv.2018.03.001.
https://doi.org/10.1016/j.jcv.2018.03.001 PMid:29525635
- Wysocki
CA (December 2017). Comparing hemophagocytic lymphohistiocytosis in
pediatric and adult patients. Current Opinion in Allergy and Clinical
Immunology. 17 (6): 405-413. doi:10.1097/ACI.0000000000000405. S2CID
11439142. https://doi.org/10.1097/ACI.0000000000000405 PMid:28957822
- Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak
CC, Gehring S, Gennery A, Gilmour KC, Gonzalez-Granado LI,
Groß-Wieltsch U, Ifversen M, Lingman-Framme J, Matthes-Martin S,
Mesters R, Meyts I, van Montfrans JM, Pachlopnik Schmid J, Pai SY,
Soler-Palacin P, Schuermann U, Schuster V, Seidel MG, Speckmann C,
Stepensky P, Sykora KW, Tesi B, Vraetz T, Waruiru C, Bryceson YT,
Moshous D, Lehmberg K, Jordan MB, Ehl S (July 2015). The syndrome of
hemophagocytic lymphohistiocytosis in primary immunodeficiencies:
implications for differential diagnosis and pathogenesis.
Haematologica. 100 (7): 978-88. doi:10.3324/haematol.2014.121608.
https://doi.org/10.3324/haematol.2014.121608 PMid:26022711
PMCid:PMC4486233
- Daver N, McClain K, Allen CE,
Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M,
Jordan MB, La Rosée P, Kantarjian HM (September 2017). A consensus
review on malignancy-associated hemophagocytic lymphohistiocytosis in
adults. Cancer. 123 (17): 3229-3240. doi:10.1002/cncr.30826.
https://doi.org/10.1002/cncr.30826 PMid:28621800 PMCid:PMC5568927
- Marsh
RA (2017). Epstein-Barr Virus and Hemophagocytic Lymphohistiocytosis.
Frontiers in Immunology. 8: 1902. doi:10.3389/fimmu.2017.01902.
https://doi.org/10.3389/fimmu.2017.01902 PMid:29358936 PMCid:PMC5766650
- Wang Y, Wang Z (January 2017). Treatment of
hemophagocytic lymphohistiocytosis. Current Opinion in Hematology. 24
(1): 54-58. doi:10.1097/MOH.0000000000000302. S2CID 31318625.
https://doi.org/10.1097/MOH.0000000000000302 PMid:27755125
- Cohen
JI, Jafe ES, Dale JK et al (2011). Characterization and treatment of
chronic active Epstein-Barr virus disease: a 28-year experience in the
United States. Blood 117(22):5835-5849
https://doi.org/10.1182/blood-2010-11-316745 PMid:21454450
PMCid:PMC3112034
- Kimura H, Ito Y, Kawabe S et al
(2012). EBV-associated T/NK-cell lymphoproliferative diseases in
nonimmunocompromised hosts: prospective analysis of 108 cases. Blood
119(3):673-686 https://doi.org/10.1182/blood-2011-10-381921
PMid:22096243
- Okano M, Kawa K, Kimura H et al
(2005). Proposed guidelines for diagnosing chronic active Epstein-Barr
virus infection. Am J Hematol 80(1):64-69
https://doi.org/10.1002/ajh.20398 PMid:16138335
- Plaza
JA, Sangueza M (2015). Hydroa vacciniforme-like lymphoma with primarily
periorbital swelling: 7 cases of an atypical clinical manifestation of
this rare cutaneous T-cell lymphoma. Am J Dermatopathol 37(1):20-25
https://doi.org/10.1097/DAD.0000000000000158 PMid:25162933
- Bollard
CM, Cohen JI (2018). How I treat T-cell chronic active Epstein-Barr
virus disease. Blood 131(26):2899-2905
https://doi.org/10.1182/blood-2018-03-785931 PMid:29712633
PMCid:PMC6024635
- Cohen JI, Manoli I, Dowdell K et
al (2019). Hydroa vacciniformelike lymphoproliferative disorder: an EBV
disease with a low risk of systemic illness in whites. Blood
133(26):2753-2764 https://doi.org/10.1182/blood.2018893750
PMid:31064750 PMCid:PMC6598378
- Okuno Y, Murata T,
Sato Y et al (2019). Defective Epstein-Barr virus in chronic active
infection and haematological malignancy. Nat Microbiol 4(3):404-413
https://doi.org/10.1038/s41564-018-0334-0 PMid:30664667
- Chiu
TM, Lin YM, Wang SC, Tsai YG (August 2016). Hypersensitivity to
mosquito bites as the primary clinical manifestation of an Epstein-Barr
virus infection. Journal of Microbiology, Immunology, and Infection =
Wei Mian Yu Gan Ran Za Zhi. 49 (4): 613-6.
doi:10.1016/j.jmii.2014.01.008.
https://doi.org/10.1016/j.jmii.2014.01.008 PMid:24662020
- Ennishi
D. The biology of the tumor microenvironment in DLBCL: Targeting the
"don't eat me" signal. J Clin Exp Hematop. 2021 Dec 22;61(4):210-215.
doi: 10.3960/jslrt.21015. Epub 2021 Sep 10.
https://doi.org/10.3960/jslrt.21015 PMid:34511583 PMCid:PMC8808113
- Sawada A, Inoue M, Kawa K (April 2017). How we
treat chronic active Epstein-Barr virus infection. International
Journal of Hematology. 105 (4): 406-418. doi:10.1007/s12185-017-2192-6.
S2CID 35297787. https://doi.org/10.1007/s12185-017-2192-6 PMid:28210942
- Park S, Ko YH (January 2014). Epstein-Barr
virus-associated T/natural killer-cell lymphoproliferative disorders.
The Journal of Dermatology. 41 (1): 29-39. doi:10.1111/1346-8138.12322.
S2CID 42534926. https://doi.org/10.1111/1346-8138.12322 PMid:24438142
- Chen BJ, Chapuy B, Ouyang J et al (2013). PD-L1
expression is characteristic of a subset of aggressive B-cell lymphomas
and virus-associated malignancies. Clin Cancer Res 19(13):3462-3473
https://doi.org/10.1158/1078-0432.CCR-13-0855 PMid:23674495
PMCid:PMC4102335
- Quintanilla-Martinez L, Ko YH,
Kimura H, Jafe ES (2017). EBVpositive T-cell and NK-cell
lymphoproliferative diseases of childhood. In: Swerdlow SH, Campo E,
Harris NL, et al., eds. WHO Classifcation of Tumours of Haematopoietic
and lymphoid tissues: IARC 355-363
- Yamaguchi M,
Miyazaki K (December 2017). Current treatment approaches for NK/T-cell
lymphoma. Journal of Clinical and Experimental Hematopathology. 57
(3): 98-108. doi:10.3960/jslrt.17018.
https://doi.org/10.3960/jslrt.17018 PMid:28679966 PMCid:PMC6144191
- Montes-Mojarro
IA, Chen BJ, Ramirez-Ibarguen AF et al (2020). Mutational profle and EBV
strains of extranodal NK/T-cell lymphoma, nasal type in Latin America.
Mod Pathol 33(5):781-791 https://doi.org/10.1038/s41379-019-0415-5
PMid:31822801
- Jiang L, Gu ZH, Yan ZX et al (2015). Exome
sequencing identifes somatic mutations of DDX3X in natural
killer/T-cell lymphoma. Nat Genet 47(9):1061-1066
https://doi.org/10.1038/ng.3358 PMid:26192917
- Lee
S, Park HY, Kang SY et al (2015). Genetic alterations of JAK/ STAT
cascade and histone modifcation in extranodal NK/T-cell lymphoma nasal
type. Oncotarget 6(19):17764-17776
https://doi.org/10.18632/oncotarget.3776 PMid:25980440
PMCid:PMC4627344
- Xiong J, Cui BW, Wang N et al
(2020). Genomic and transcriptomic characterization of natural killer T
cell lymphoma. Cancer Cell 37(3):403-419 e406
https://doi.org/10.1016/j.ccell.2020.02.005 PMid:32183952
- Dong
G, Liu X, Wang L et al (2022). Genomic profling identifes distinct
genetic subtypes in extra-nodal natural killer/T-cell lymphoma.
Leukemia 36(8):2064-2075. https://doi.org/10.1038/s41375-022-01623-z
PMid:35697790 PMCid:PMC10499270
- Broccoli A, Zinzani PL
(March 2017).. Peripheral T-cell lymphoma, not otherwise specified.
Blood. 129 (9): 1103-1112. doi:10.1182/blood-2016-08-692566.
https://doi.org/10.1182/blood-2016-08-692566 PMid:28115372
- Nemani
S, Korula A, Agrawal B, Kavitha ML, Manipadam MT, Sigamani E, George B,
Srivastava A, Viswabandya A, Mathews V (May 2018). Peripheral T cell
lymphoma: Clinico-pathological characteristics & outcome from a
tertiary care centre in south India. The Indian Journal of Medical
Research. 147 (5): 464-470. doi:10.4103/ijmr.IJMR_1108_16.
https://doi.org/10.4103/ijmr.IJMR_1108_16 PMid:30082570
PMCid:PMC6094517
- Khan N, Ozkaya N, Moskowitz A,
Dogan A, Horwitz S (September 2018). Peripheral T-cell lymphoma-are we
making progress?. Best Practice & Research. Clinical Haematology.
31 (3): 306-314. doi:10.1016/j.beha.2018.07.010. PMC 8941989. S2CID
52273670. https://doi.org/10.1016/j.beha.2018.07.010 PMid:30213401
PMCid:PMC8941989
- Ludvigsen M, Bjerregård Pedersen
M, Lystlund Lauridsen K, Svenstrup Poulsen T, Hamilton-Dutoit SJ,
Besenbacher S, Bendix K, Møller MB, Nørgaard P, d'Amore F, Honoré B
(October 2018). Proteomic profiling identifies outcome-predictive
markers in patients with peripheral T-cell lymphoma, not otherwise
specified. Blood Advances. 2 (19): 2533-2542.
doi:10.1182/bloodadvances.2018019893.
https://doi.org/10.1182/bloodadvances.2018019893 PMid:30291111
PMCid:PMC6177647
- Mosalpuria K, Bociek RG, Vose JM
(January 2014). Angioimmunoblastic T-cell lymphoma management.
Seminars in Hematology. 51 (1): 52-8.
doi:10.1053/j.seminhematol.2013.11.008.
https://doi.org/10.1053/j.seminhematol.2013.11.008 PMid:24468316
- Hu
S, Young KH, Konoplev SN, Medeiros LJ (November 2012). Follicular
T-cell lymphoma: a member of an emerging family of follicular helper
T-cell derived T-cell lymphomas. Human Pathology. 43 (11): 1789-98.
doi:10.1016/j.humpath.2012.05.002.
https://doi.org/10.1016/j.humpath.2012.05.002 PMid:22959759
- Zhang
H, Meng Q, Yin W, Xu L, Lie L (July 2013). Adult aggressive natural
killer cell leukemia. The American Journal of the Medical Sciences.
346 (1): 56-63. doi:10.1097/MAJ.0b013e3182764b59. S2CID 32910828.
https://doi.org/10.1097/MAJ.0b013e3182764b59 PMid:23241562
- Lima
M (October 2015). Extranodal NK/T cell lymphoma and aggressive NK cell
leukaemia: evidence for their origin on CD56+bright CD16-/+dim NK
cells. Pathology. 47 (6): 503-14. doi:10.1097/PAT.0000000000000275.
S2CID 5264015. https://doi.org/10.1097/PAT.0000000000000275
PMid:26166665
- Tang YT, Wang D, Luo H, Xiao M, Zhou HS,
Liu D, Ling SP, Wang N, Hu XL, Luo Y, Mao X, Ao QL, Huang J, Zhang W,
Sheng LS, Zhu LJ, Shang Z, Gao LL, Zhang PL, Zhou M, Zhou KG, Qiu LG,
Liu QF, Zhang HY, Li JY, Jin J, Fu L, Zhao WL, Chen JP, Du X, Huang G,
Wang QF, Zhou JF, Huang L (December 2017). Aggressive NK-cell
leukemia: clinical subtypes, molecular features, and treatment
outcomes. Blood Cancer Journal. 7 (12): 660.
doi:10.1038/s41408-017-0021-z.
https://doi.org/10.1038/s41408-017-0021-z PMid:29263371
PMCid:PMC5802497
- Bi Y, Huo Z, Liang Z, Meng Y, Jia C,
Shi X, Song L, Luo Y, Ling Q, Liu T (July 2015). Intravascular NK-cell
lymphoma: a case report and review of the literature. Diagnostic
Pathology. 10: 84. doi:10.1186/s13000-015-0336-7.
https://doi.org/10.1186/s13000-015-0336-7 PMid:26126576
PMCid:PMC4488042
- Yan J, Zhang F, Luo D, Yao S, Chen Y,
Xu F, Luo X, He J, Liu Y (2017). Intravascular NK/T-cell lymphoma: a
series of four cases. International Journal of Clinical and
Experimental Pathology. 10 (9): 9541-9550. PMC 6965900 PMID 31966830.
- Zanelli
M, Mengoli MC, Del Sordo R, Cagini A, De Marco L, Simonetti E, Martino
G, Zizzo M, Ascani S (November 2018). Intravascular NK/T-cell
lymphoma, Epstein-Barr virus positive with multiorgan involvement: a
clinical dilemma. BMC Cancer. 18 (1): 1115.
doi:10.1186/s12885-018-5001-6.
https://doi.org/10.1186/s12885-018-5001-6 PMid:30442097
PMCid:PMC6238309
- Gleason BC, Brinster NK, Granter SR,
Pinkus GS, Lindeman NI, Miller DM (February 2008). Intravascular
cytotoxic T-cell lymphoma: A case report and review of the literature.
Journal of the American Academy of Dermatology. 58 (2): 290-4.
doi:10.1016/j.jaad.2006.12.022.
https://doi.org/10.1016/j.jaad.2006.12.022 PMid:18222325
- Wang
L, Chen S, Ma H, Shi D, Huang C, Lu C, Gao T, Wang G (September
2015). Intravascular NK/T-cell lymphoma: a report of five cases
with
cutaneous manifestation from China. Journal of Cutaneous Pathology. 42
(9): 610-7. doi:10.1111/cup.12515. S2CID 23046075.
https://doi.org/10.1111/cup.12515 PMid:25931234
- Melchers
RC, Willemze R, Jansen PM, Daniëls LA, Vermeer MH, Quint KD (June
2019). A rare case of cutaneous Epstein-Barr virus-negative
intravascular cytotoxic T-cell lymphoma. JAAD Case Reports. 5 (6):
548-551. doi:10.1016/j.jdcr.2019.04.013.
https://doi.org/10.1016/j.jdcr.2019.04.013 PMid:31245517
PMCid:PMC6581970
- Campo E, Jafe ES, Cook JR et al
(2022). The international consensus classifcation of mature lymphoid
neoplasms: a report from the clinical advisory committee. Blood
140(11):1229-1253. https://doi.org/10.1182/blood.2022015851
PMid:35653592 PMCid:PMC9479027
- Allen UD, Preiksaitis JK
(2019). Practice ASTIDCo Post-transplant lymphoproliferative disorders,
Epstein-Barr virus infection, and disease in solid organ
transplantation: guidelines from the American Society of
Transplantation Infectious Diseases Community of Practice. Clin
Transplant 33(9):e13652 https://doi.org/10.1111/ctr.13652 PMid:31230381
- Shah N, Eyre TA, Tucker D et al (2021). Front-line
management of post-transplantation lymphoproliferative disorder in
adult solid organ recipient patients - a British Society for
Haematology Guideline. Br J Haematol 193(4):727-740
https://doi.org/10.1111/bjh.17421 PMid:33877688
- Vakiani
E, Nandula SV, Subramaniyam S et al (2007). Cytogenetic analysis of
B-cell posttransplant lymphoproliferations validates the World Health
Organization classifcation and suggests inclusion of forid follicular
hyperplasia as a precursor lesion. Hum Pathol 38(2):315-325
https://doi.org/10.1016/j.humpath.2006.08.014 PMid:17134734
- Butzmann
A, Sridhar K, Jangam D et al (2021). Mutations in JAK/ STAT and NOTCH1
genes are enriched in post-transplant lymphoproliferative disorders.
Front Oncol 11:790481 https://doi.org/10.3389/fonc.2021.790481
PMid:35111674 PMCid:PMC8801788
- Marcelis L,
Tousseyn T (2019). The tumor microenvironment in post-transplant
lymphoproliferative disorders. Cancer Microenviron. 12(1):3-16
https://doi.org/10.1007/s12307-018-00219-5 PMid:30680693
PMCid:PMC6529504
- Nelson AA, Harrington AM, Kroft
S, Dahar MA, Hamadani M, Dhakal B (2016). Presentation and management of
post-allogeneic transplantation EBV-positive mucocutaneous ulcer. Bone
Marrow Transplant 51(2):300-302 https://doi.org/10.1038/bmt.2015.245
PMid:26457913
- Menter T, Juskevicius D, Alikian M
et al (2017). Mutational landscape of B-cell post-transplant
lymphoproliferative disorders. Br J Haematol 178(1):48-56
https://doi.org/10.1111/bjh.14633 PMid:28419429
- King
RL, Khurana A, Mwangi R et al (2021). Clinicopathologic characteristics,
treatment, and outcomes of post-transplant lymphoproliferative
disorders: a single-institution experience using 2017 WHO diagnostic
criteria. Hemasphere 5(10):e640
https://doi.org/10.1097/HS9.0000000000000640 PMid:34514344
PMCid:PMC8423401
- Galera P, Flavin R, Savage NM et
al (2020). Epstein-Barr virusnegative marginal zone lymphoma as an
uncommon form of monomorphic posttransplant lymphoproliferative
disorder. Am J Surg Pathol 44(10):1340-1352
https://doi.org/10.1097/PAS.0000000000001514 PMid:32554995
PMCid:PMC7492423
- Morscio J, Tousseyn T (2016). Recent
insights in the pathogenesis of post-transplantation
lymphoproliferative disorders. World J Transplant 6(3):505-516
https://doi.org/10.5500/wjt.v6.i3.505 PMid:27683629 PMCid:PMC5036120
- Margolskee E, Jobanputra V, Jain P et al (2016).
Genetic landscape of T- and NK-cell post-transplant lymphoproliferative
disorders. Oncotarget 7(25):37636-37648
https://doi.org/10.18632/oncotarget.9400 PMid:27203213 PMCid:PMC5122338
- Bobillo S, Abrisqueta P, Sanchez-Gonzalez B et al
(2018). Posttransplant monomorphic Burkitt's lymphoma: clinical
characteristics and outcome of a multicenter series. Ann Hematol
97(12):2417-2424 https://doi.org/10.1007/s00277-018-3473-8
PMid:30116871
- Ferreiro JF, Morscio J, Dierickx D et al
(2015). Post-transplant molecularly defined Burkitt lymphomas are
frequently MYCnegative and characterized by the 11q-gain/loss pattern.
Haematologica 100(7):e275-279
https://doi.org/10.3324/haematol.2015.124305 PMid:25795716
PMCid:PMC4486241
- Kaji D, Kusakabe M, Sakata-Yanagimoto
M et al (2021). Retrospective analyses of other iatrogenic
immunodefciency-associated lymphoproliferative disorders in patients
with rheumatic diseases. Br J Haematol 195(4):585-594
https://doi.org/10.1111/bjh.17824 PMid:34558064 PMCid:PMC9290981
- Goodlad
JR (June 2017). Epstein-Barr virus-associated Lymphoproliferative
Disorders in the Skin. Surgical Pathology Clinics. 10 (2): 429-453.
doi:10.1016/j.path.2017.01.001.
https://doi.org/10.1016/j.path.2017.01.001 PMid:28477890
- Casulo
C, Friedberg J (September 2015). Treating Burkitt Lymphoma in Adults.
Current Hematologic Malignancy Reports. 10 (3): 266-71.
doi:10.1007/s11899-015-0263-4. S2CID 21258747.
https://doi.org/10.1007/s11899-015-0263-4 PMid:26013028
- Molyneux
EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, Harrison CJ,
Israels T, Bailey S (March 2012). Burkitt's lymphoma (PDF). Lancet.
379 (9822): 1234-44. doi:10.1016/S0140-6736(11)61177-X. S2CID 39960470.
https://doi.org/10.1016/S0140-6736(11)61177-X PMid:22333947
- Vockerodt
M, Yap LF, Shannon-Lowe C, Curley H, Wei W, Vrzalikova K, Murray PG
(January 2015). The Epstein-Barr virus and the pathogenesis of
lymphoma. The Journal of Pathology. 235 (2): 312-22.
doi:10.1002/path.4459. S2CID 22313509.
https://doi.org/10.1002/path.4459 PMid:25294567
- Navari
M, Etebari M, De Falco G, Ambrosio MR, Gibellini D, Leoncini L,
Piccaluga PP (2015). The presence of Epstein-Barr virus significantly
impacts the transcriptional profile in immunodeficiency-associated
Burkitt lymphoma. Frontiers in Microbiology. 6: 556.
doi:10.3389/fmicb.2015.00556. https://doi.org/10.3389/fmicb.2015.00556
PMid:26113842 PMCid:PMC4462103
- Kaplan LD (March
2012). HIV-associated lymphoma. Best Practice & Research.
Clinical Haematology. 25 (1): 101-17. doi:10.1016/j.beha.2012.01.001.
https://doi.org/10.1016/j.beha.2012.01.001 PMid:22409827
- Tang
VK, Vijhani P, Cherian SV, Ambelil M, Estrada-Y-Martin RM
(2018). Primary pulmonary lymphoproliferative neoplasms. Lung
India. 35 (3):
220-230. doi:10.4103/lungindia.lungindia_381_17.
https://doi.org/10.4103/lungindia.lungindia_381_17 PMid:29697079
PMCid:PMC5946555
- Chavez JC, Sandoval-Sus J, Horna P,
Dalia S, Bello C, Chevernick P, Sotomayor EM, Sokol L, Shah B (August
2016). Lymphomatoid Granulomatosis: A Single Institution Experience
and Review of the Literature. Clinical Lymphoma, Myeloma &
Leukemia. 16 Suppl: S170-4. doi:10.1016/j.clml.2016.02.024.
https://doi.org/10.1016/j.clml.2016.02.024 PMid:27521314
- Gangar
P, Venkatarajan S (July 2015). Granulomatous Lymphoproliferative
Disorders: Granulomatous Slack Skin and Lymphomatoid Granulomatosis.
Dermatologic Clinics. 33 (3): 489-96. doi:10.1016/j.det.2015.03.013.
https://doi.org/10.1016/j.det.2015.03.013 PMid:26143428
- Sigamani
E, Chandramohan J, Nair S, Chacko G, Thomas M, Mathew LG, Pulimood S,
Manipadam MT (2018). Lymphomatoid granulomatosis: A case series from
South India. Indian Journal of Pathology & Microbiology. 61 (2):
228-232. doi:10.4103/IJPM.IJPM_471_17.
https://doi.org/10.4103/IJPM.IJPM_471_17 PMid:29676363
- Shannon-Lowe
C, Rickinson AB, Bell AI (October 2017). Epstein-Barr virus-associated
lymphomas. Philosophical Transactions of the Royal Society of London.
Series B, Biological Sciences. 372 (1732): 20160271.
doi:10.1098/rstb.2016.0271. https://doi.org/10.1098/rstb.2016.0271
PMid:28893938 PMCid:PMC5597738
- Li S, Young KH,
Medeiros LJ (January 2018). Diffuse large B-cell lymphoma. Pathology.
50 (1): 74-87. doi:10.1016/j.pathol.2017.09.006. S2CID
20839613.[permanent dead link]
https://doi.org/10.1016/j.pathol.2017.09.006 PMid:29167021
- Jain
N, Keating MJ (August 2016). Richter transformation of CLL. Expert
Review of Hematology. 9 (8): 793-801.
doi:10.1080/17474086.2016.1199948. S2CID 23968856.
https://doi.org/10.1080/17474086.2016.1199948 PMid:27351634
- Grimm
KE, O'Malley DP (October 2018). Aggressive B cell lymphomas in the
2017 revised WHO classification of tumors of hematopoietic and lymphoid
tissues. Annals of Diagnostic Pathology. 38: 6-10.
doi:10.1016/j.anndiagpath.2018.09.014. S2CID 53196244.
https://doi.org/10.1016/j.anndiagpath.2018.09.014 PMid:30380402
- Aozasa
K (March 2006). Pyothorax-associated lymphoma. Journal of Clinical
and Experimental Hematopathology. 46 (1): 5-10. doi:10.3960/jslrt.46.5.
https://doi.org/10.3960/jslrt.46.5 PMid:17058803
- Boyer
DF, McKelvie PA, de Leval L, Edlefsen KL, Ko YH, Aberman ZA, Kovach AE,
Masih A, Nishino HT, Weiss LM, Meeker AK, Nardi V, Palisoc M, Shao L,
Pittaluga S, Ferry JA, Harris NL, Sohani AR (March 2017). Fibrin-associated EBV-positive Large B-Cell Lymphoma: An Indolent
Neoplasm With Features Distinct From Diffuse Large B-Cell Lymphoma
Associated With Chronic Inflammation. The American Journal of Surgical
Pathology. 41 (3): 299-312. doi:10.1097/PAS.0000000000000775. S2CID
3521190. https://doi.org/10.1097/PAS.0000000000000775 PMid:28195879
- Vets,
J., Marcelis, L., Schepers, C. et al. (2023). Breast implant associated
EBV-positive Diffuse Large B-cell lymphoma: an underrecognized entity?.
Diagn Pathol 18, 52 . https://doi.org/10.1186/s13000-023-01337-5
PMid:37098615 PMCid:PMC10127423
- Linke-Serinsöz E, Fend
F, Quintanilla-Martinez L (July 2017). Human immunodeficiency virus
(HIV) and Epstein-Barr virus (EBV) related lymphomas, pathology view
point. Seminars in Diagnostic Pathology. 34 (4): 352-363.
doi:10.1053/j.semdp.2017.04.003.
https://doi.org/10.1053/j.semdp.2017.04.003 PMid:28506687
- Tchernonog
E, Faurie P, Coppo P, Monjanel H, Bonnet A, Algarte Génin M, Mercier M,
Dupuis J, Bijou F, Herbaux C, Delmer A, Fabiani B, Besson C, Le Gouill
S, Gyan E, Laurent C, Ghesquieres H, Cartron G (April 2017). Clinical
characteristics and prognostic factors of plasmablastic lymphoma
patients: analysis of 135 patients from the LYSA group. Annals of
Oncology. 28 (4): 843-848. doi:10.1093/annonc/mdw684.
https://doi.org/10.1093/annonc/mdw684 PMid:28031174
- Yan
J, Wang J, Zhang W, Chen M, Chen J, Liu W (April 2017). Solitary
plasmacytoma associated with Epstein-Barr virus: a clinicopathologic,
cytogenetic study and literature review. Annals of Diagnostic
Pathology. 27: 1-6. doi:10.1016/j.anndiagpath.2016.09.002.
https://doi.org/10.1016/j.anndiagpath.2016.09.002 PMid:28325354