Ugo
Testa1, Giuseppe Leone2,
Elvira Pelosi1, Germana Castelli1
and Valerio De Stefano2,3.
1 Istituto Superiore di Sanità, Roma, Italy.
2 Section of Hematology, Department of
Radiological and Hematological Sciences, Catholic University, Rome
3 Department of Laboratory and Hematological
Sciences, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome
Published: May 01, 2024
Received: March 20, 2024
Accepted: April 16, 2024
Mediterr J Hematol Infect Dis 2024, 16(1): e2024044 DOI
10.4084/MJHID.2024.044
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
study of monoclonal serum proteins has led to the generation of two
major theories: one proposing that individuals who had monoclonal
proteins without any symptoms or evidence of end-organ damage have a
benign condition, the other one suggesting that some individuals with
asymptomatic monoclonal proteins may progress to multiple myeloma and
thus are affected by a monoclonal gammopathy of undetermined
significance (MGUS). Longitudinal studies of subjects with MGUS have
supported the second theory. Subsequent studies have characterized and
defined the existence of another precursor of multiple myeloma,
smoldering multiple myeloma (SMM), intermediate between MGUS and
multiple myeloma. Primary molecular events, chromosome translocations,
and chromosome number alterations resulting in hyperploidy, required
for multiple myeloma development, are already observed in myeloma
precursors. MGUS and SMM are heterogeneous conditions with the presence
of tumors with distinct pathogenic phenotypes and clinical outcomes.
The identification of MGUS and SMM patients with a molecularly defined
high risk of progression to MM offers the unique opportunity of early
intervention with a therapeutic approach on a low tumor burden.
|
Introduction
Multiple
myeloma (MM) is a disorder of the monoclonal plasma cells. It is the
second most common hematologic malignancy, and its incidence is
increasing. The current estimated annual incidence rate (AIR) is very
different in the various countries; high-income countries reported the
highest incidence: Australia and New Zealand with an incidence (AIR
4.86 [4.66-5.07]), Northern America (4.74 [4.69-4.79]), and northern
Europe (3.82 [3.71-3.93]) The lowest incidences were observed in
western Africa (0.81 [0.39-1.66]), Melanesia (0.87 [0.55-1.37]), and
southeastern Asia (0.96 [0.73-1.27]). In the USA, the incidence was 7.7
per 100,000 inhabitants (2019), with a 126% increase since 2000, when
the incidence was 6.1 per 100,000.[1] MM may originate
from the evolution of precursor conditions, including monoclonal
gammopathy of undetermined significance (MGUS) and smoldering multiple
myeloma (SMM).
Patients with precursors to MM are dichotomized as
having MGUS or SMM based on monoclonal protein concentrations or plasma
cell percentage in the bone marrow. The current diagnostic criteria for
MGUS imply the presence of a serum monoclonal protein (M protein) at a
concentration of <3g/dL, bone marrow with <10% monoclonal plasma
cells, and absence of end-organ damage (lytic bone lesions, anemia,
hypercalcemia, kidney impairment, hyperviscosity) related to the
proliferation of plasma cells. Diagnostic criteria for SMM imply the
presence of serum M protein (IgG or IgA) ≥ 3g/dL or urinary M protein ≥
500mg/24h and/or 10%-59% clonal plasma cells in the absence of
end-organ damage attributable to the plasma cell disorder.[1,2]
Genetic Alterations in MGUS
MGUS occurs in about 3% of individuals 50 years of age or older.[2]
This estimate was based on the current routine methodology based on
serum protein electrophoresis supplemented by immunofixation. However,
recently developed mass spectrometry (MS)-based approaches have allowed
a markedly greater sensitivity in the detection and quantification of
M-proteins, showing that the prevalence of MGUS might be two/three
times higher than previously estimated using serum protein
electrophoresis.[3-5] Interestingly, the mass
spectrometry evaluation allowed to distinguish two types of MGUS:
monoclonal gammopathies below the clinical immunofixation
electrophoresis detection level (>0.2 g/L) defined as monoclonal
gammopathy of indeterminate potential (MGIP, predominantly of
immunoglobulin M isotype); monoclonal gammopathies with higher M
protein concentrations, defined as mass- spectrometry MGUS.[5]
The prevalence of MGIP among 7622 participants increased with age: 19%]
for individuals aged <50 years, 29% for those aged ≥50 years, and
37% for 946] for those aged ≥70 years.4 However, the large discrepancy
between the prevalence of MGIP and MGUS in the general population
(particularly in older individuals) and the relative rarity of myeloma
indicates that evolution in myeloma requires very complex and subtle
rare mechanisms.
A few risk factors have been involved in MGUS
development, including age, male, sex, Black or African American race,
and family history. The definition of a category of MGUS patients with
an M protein of 0.2 g/dL and identified as MS-MGUS allows us to show an
epidemiological link between MGUS and obesity and heavy smoking.[6]
A
fundamental study by Kyle and coworkers explored the long-term
follow-up of 1384 subjects with MGUS; MGUS progression was observed in
11% of these patients; the risk of progression was estimated at 10% at
10 years, 18% at 20 years, 28% at 30 years, 36% at 35 years and 36% at
40 years.[2] Among patients with IgM MGUS, the
presence of two risk factors, such as high serum M-protein (≥1.5 g/dL)
and an abnormal serum-free light-chain ratio (ratio of kappa to lambda
free light chains), was associated with a risk of progression at 20
years of 55%, compared to 41% in those with one adverse risk factor and
19% in patients without any of the two risk factors[2] (Table 1, Figure 1).
Among patients with non-IgM MGUS, the presence of two risk factors was
associated with a risk of progression at 20 years of 30%, 20% among
those with one risk factor and 7% in those without neither risk factor.[2]
Importantly, individuals with MGUS have a shorter survival rate than
those without MGUS in a control population matched for age and sex.[2]
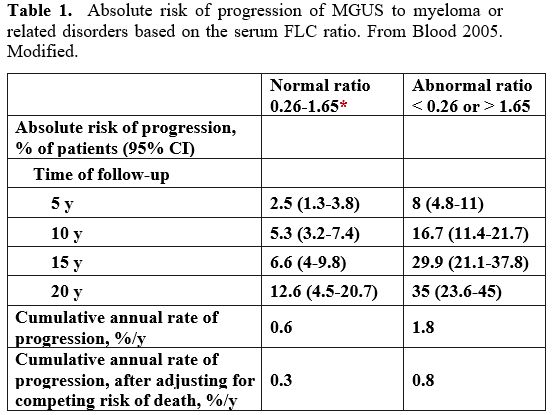 |
Table 1. Absolute risk of
progression of MGUS to myeloma or related disorders based on the serum
FLC ratio. From Blood 2005. Modified. |
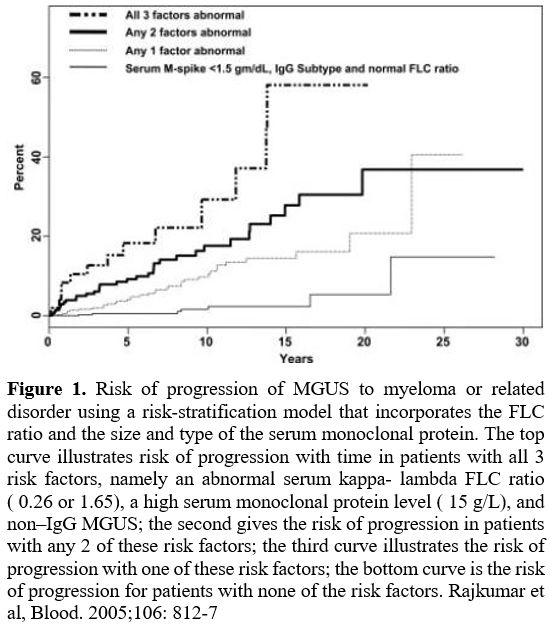 |
Figure
1. Risk of progression of MGUS to myeloma or related disorder using a
risk-stratification model that incorporates the FLC ratio and the size
and type of the serum monoclonal protein. The top curve illustrates
risk of progression with time in patients with all 3 risk factors,
namely an abnormal serum kappa- lambda FLC ratio ( 0.26 or 1.65), a
high serum monoclonal protein level ( 15 g/L), and non–IgG MGUS; the
second gives the risk of progression in patients with any 2 of these
risk factors; the third curve illustrates the risk of progression with
one of these risk factors; the bottom curve is the risk of progression
for patients with none of the risk factors. Rajkumar et al, Blood.
2005;106: 812-7
|
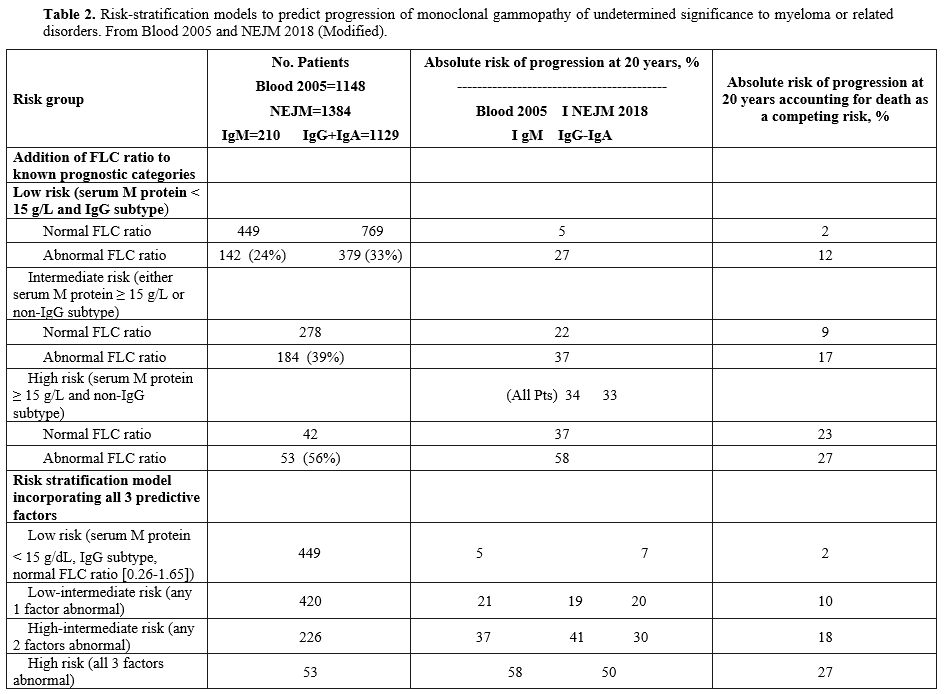 |
Table 2.
Risk-stratification models to predict progression of monoclonal
gammopathy of undetermined significance to myeloma or related
disorders. From Blood 2005 and NEJM 2018 (Modified).
|
It
is of interest to note that there are three different asymptomatic
conditions characterized by clonal expansion of blood cells: MGUS,
monoclonal B-cell lymphocytosis (MBL), and clonal hematopoiesis (CH).
All these three conditions are associated with an increased risk of
hematologic cancers; particularly, each condition has an annual
progression rate of about 1-2% per year, with MGUS progressing to MM,
MBL to chronic lymphocytic leukemia, and CH to myeloid neoplasia.
Furthermore, all three premalignant conditions are associated with
adverse outcomes. A common feature of all these states is their
consistent heterogeneity at the mutational level, including a set of
gene abnormalities acquired by apparently stochastic processes, driving
changes in biological behavior, and generation of multiple clonal
propagating units in the competition. Screening on non-hematological
patients showed that there is no association between these three
premalignant conditions, thus supporting their independent origin.[7]
Initial
oncogenic events commonly displayed by MGUS and MM are characterized by
at least one of seven primary immunoglobulin heavy chain gene
translocations at q32 or by hyperploidy (about 50% of cases) related to
trisomy of several chromosomes (3, 5, 7, 9, 11, 15, 19 and 21).[8-10]
Dysregulation of the G1-S cell-cycle transition through overexpression
of the cyclin D gene is an event observed in both non-hyperploid and
hyperploid MGUS; it is an early event in MM development.[8]
The
analysis of the clonality of copy number alterations (CNAs), including
those related to whole chromosomes or segments of chromosomes, was
carried out by Samur et al. in 164 samples.[11] 30.5%
of the MGUS were classified as hyperploid, and in these tumors, gains
in chromosomes 19 (95%), 15 (86%), and 9 (87%) were the most frequent
events; in the non-hyperploid group, del 13 was the most frequent event
(21%), followed by the gain of 1q (13%).[11]
Importantly, the most recurrent CNAs observed in hyperploid MGUS are
also observed in hyperploid MM, thus confirming the occurrence of these
events very early in the disease process, while the majority of
subclonal deletions (deletions targeting 1q, 6q, 8p, 12p, 12q, 14q,
16p, 16q and 17p) detected in MM patients, were not observed in MGUS
patients, thus suggesting that they are late events.[11]
CNAs
are frequently observed in MGUS patients, and their number is lower
than that observed in MM; furthermore, the number of CNAs is higher in
MGUS patients who progress to MM compared to those who did not progress
to MM.[12]
Amplification of the chromosomal
region 1q21 is the most recurrent chromosomal gain observed in MM; its
frequency is higher in MM (40%) than in MGUS (25%) patients, and its
presence is associated with a higher risk of progression of MGUS to MM.[13] Several candidate oncogenes are contained in region 1q21, and one of them, ILF2, plays a relevant role in MM development, progression, and drug resistance.[14]
ILF2 promotes its oncogenic effects in MM cells through interaction
with APOBECC3B, potentiating its DNA cytosine deaminase activity, thus
favoring DNA genomic instability.[15] 1q21 gain/amplification has a negative prognostic value.[16]
The frequency of 1p deletions is much lower in MGUS (about 5%) than in MM (about 30%).[11] This deletion implies the loss of two tumor suppressor genes, CDKN2C and FAM46C.[17] Particularly, the deletion of 1p32.3, which involves loss of CDKN2C, is associated with adverse overall survival.[18]
Complete
loss of chromosome 13 is more frequent in MM than in MGUS patients.
However, the frequency of chromosome loss in MGUS is associated with
the presence of some specific IgH translocations, such as t(4;14) and
t(14;16) translocations, but absent in other IgH translocations, such
as t(6;14) and t(11;14)(16).[19]
The clonality analysis of the CNAs in MGUS was carried out by Samur et al. in 164 samples.[11]
30.5% of MGUS were classified as hyperdiploid, and in these tumors,
gains in chromosomes 19 (95%), 15 (86%), and 9 (87%) are the most
frequent events; in the nonhyperdiploid group, del 13 was the most
frequent event (21%), followed by the gain of 1q (13%).[11]
Importantly, the CNAs observed in hyperdiploid MGUS are also observed
in hyperdiploid MM, thus confirming the occurrence of these events very
early in the disease process; in contrast the majority of subclonal
deletions (deletions targeting 1q, 6q, 8p, 12p, 12q, 14q, 16p, 16q and
17p) detected in MM patients, were not observed in MGUS patients, thus
suggesting that they are late events.[11]
Whole
exome sequencing studies have shown the presence of non-synonymous
mutations and copy number alterations in 97% and 60% of MGUS cases,
respectively; somatic mutations in MGUS were markedly less frequent
than in MM.[20] Few genes were similarly mutated in MGUS and MM; IGH translocations are present in similar frequency in MGUS and MM; MYC translocations and TP53 mutations are not observed in MGUS, thus indicating that these alterations are drivers of progression to MM.[20]
Studies
of characterization of molecular alterations of MGUS and MM suggest a
classification of MGUS into monoclonal gammopathy and early multiple
myeloma (eMM): monoclonal gammopathy is characterized by the presence
of canonical IGH translocations and hyperploidy, while additional
genetic abnormalities are observed in eMM and MM, such as mutations in
driver genes, copy number alterations, MYC translocation, complex genetic events.[21]
MGUSs classified as monoclonal gammopathy have a low risk of
progression to MM, while those classified as eMM have a high risk of MM
progression.[21] These conclusions were supported by
whole genome sequencing studies of MGUS, SMM, and MM, showing that
cases with a non-progressing, clinically stable myeloma precursor
condition are characterized by later initiation in the life of patients
and by the absence of myeloma-defining genomic events, including
chromotripsis, templated insertion, mutations in driver genes, and
canonical APOBEC mutational activity.[22]
Particularly in stable myeloma precursor condition, the tumor
mutational burden, as well as the prevalence of structural variants and
copy number alterations [such as del(14q), del(16q), del(17p),
del(1p12), amp(1q24), del(6q25), del(8p), amp(8q24)] are observed at a
significantly lower number compared with progressive myeloma precursor
condition.[20] None of the stable myeloma precursor condition cases displayed any structural variant involving the MYC hotspot.[22]
The
molecular analysis of IgM MGUS and Waldenstrom macroglobulinemia (WM)
showed a similar mutational profile, with quantitative differences in
the mutational frequencies higher in WM than in IgM MGUS.[23] MYD88 was the gene most frequently mutated in both WM (85%) and IgM MGUS (47%).[21] The somatic MYD88L265P mutation determines the constitutive activation of NF-kB and stimulation of B-lymphoid proliferation. The MYD88
mutation is an early event during WM development, as supported by its
high frequency in IgM MGUS patients. The presence of MYD88 mutations
and high serum M-protein concentration (1g/dL or higher) identified a
subpopulation of high-risk IgM MGUS patients, with a 38% risk of
transformation at 10 years.[24]
IgM MGUS is a
premalignant condition for Waldenstrom macroglobulinemia and other
B-cell malignancies and very rarely for MM. It is defined by the
presence of a monoclonal protein at a level below 3g/dL with
plasmocytic bone marrow infiltration below 10%.[25]
The
gene encoding the chemokine receptor CXCR4, involved in the homing of
B-lymphoid cells in the bone marrow, is mutated in a minority of IgM
MGUS (5-10%), compared to a higher frequency of mutations observed in
WM (20-25%).[26] CXCR4 mutation is usually a subclonal event and occurs late during WM development.[26]
Moreno and coworkers have investigated MYD88 and CXCR4 mutations
by droplet digital polymerase chain reaction (ddPCR) in 101 IgM MGUS
and 69 SWM (smoldering Waldenstrom macroglobulinemia).[27] Importantly, ddPCR was more sensitive than standard PCR for the detection of MYD88L265P mutations in both IgM MGUS (64% vs. 39%) and in SWM (82% vs. 73%); the MYD88 mutation burden was markedly higher in SWM (5.36%) and WM (11%) than in IgM MGUS (1.13%); the MYD88
mutation burden correlated with the serum M-protein size, the serum IgM
concentration, the infiltration of the BM by histological evaluation of
the percentage of BM clonal B-cells by flow cytometry.[25] The two most frequent CXCR4 mutations were C1013G and C1013A; CXCR4 C1013G was positive in 35% and 43% of patients with IgM MGUS and SWM, respectively; the median CXCR4 C1013G mutation distribution in both IgM MGUS and SWM was 0.4% and suggested a subclonal pattern for CXCR4 mutations; CXCR4 C1013A mutation was more rarely observed (2/54 IgM MGUS and 3/42 SWM).[27]
Several
biological features of MGUS are helpful in stratifying the risk for
progression of MGUS to symptomatic disease. Among them, the most
relevant is represented by the size of the BM plasma cell clone and
M-protein levels. Several risk stratification models predicting MGUS
progression to MM have been proposed; these models take into account
serum M-protein levels (>15g/L), aberrant phenotype in >95% BM
plasma cells, non-IgG subtype and abnormal FLC (free light chains)
ratio as predictive of MGUS progression risk factors.[28]
Mayo Clinic MGUS is one of the most adopted risk stratification models
and implies the stratification into low, low-intermediate,
high-intermediate, and high with increasing absolute risk of
progression at 20 years.[29] Although these
prognostic models have proven their utility, they have not been useful
for identifying cases with MGUS with low- and intermediate-risk who may
have undergone malignant transformation.
MM development is
characterized by progressive stromal alterations mainly characterized
by reduced hematopoietic support, decreased osteoblast differentiation
and function, and increased osteoclast activity. A recent study showed
that abnormalities of stromal cells already occur in MGUS, such as the
presence of a high number of senescent cells and a reduced osteogenic
differentiation capacity and hematopoietic support.[30]
Furthermore, RNA sequencing studies have shown the expression of a
broad spectrum of differentially expressed genes, including genes of
the BMP/TGF-signaling pathway, present in MGUS and increasing in SMM
and MM.[30]
Transition from MGUS to SMM and MM
Several
studies have attempted to define the molecular changes that drive the
transition from MGUS to SMM and from SMM to MM. Comparisons of unpaired
MGUS/SMM and MGUS/MM samples have shown that MGUS and SMM display a
consistent similarity with MM; however, many mutations are present in a
lower proportion of malignant plasma cells.[19,31]
Thus, Lopez-Corral, using FISH, observed that the proportion of plasma
cells bearing IgH translocations, t(11;14), and 13q deletions was
significantly lower in MGUS than in MM.[28]
Furthermore, the same authors showed a progressive increase in the
incidence of CNAs from MGUS to SMM and MM (median 5, 7.5, and 12 per
case, respectively). Furthermore, it was shown that CNAs, such as 11q
and 21q gains together with 16q and 22q deletions, apparently exclusive
on MM cases, are, in fact, found as minor subclones in MGUS.[31]
In
agreement with these findings, paired-sample studies based on the
analysis of a few patients evaluating the evolution of genetic
abnormalities in the transition from MGUS to SMM[10] or from high-risk SMM to MM[32]
have identified most genetic abnormalities required for these tumor
evolutions in the premalignant stages, with the clinically dominant
subclone already present in SMM.
The ensemble of these studies
suggested that intraclonal heterogeneity is an early event in the
development and occurs at stages anterior to MM. Whole exome sequencing
studies of five paired cases with the evolution from MGUS to SMM and
five with the evolution from SMM to MM further supported this model of
MM development, showing that MM development is mainly characterized by
the phenomenon of clonal stability, with the highly transformed
subclonal populations observed in MM being already present at the
stages of precursor lesions (MGUS and SMM).[33]
Bolli
et al. reported the analysis of 10 SMM patients progressing to MM by
whole-genome analysis of 10 paired SMM and MM samples; the analysis of
the genomic landscape, including mutational profile and structural
rearrangements, showed a similarity between the SMM stage and the MM
stage.[34] Paired sample analysis showed two
different patterns of progression: 60% of SMM patients evolved
according to a spontaneous evolution process implying a change in
subclonal composition from SMM to MM in a branching pattern, reflecting
a spontaneous evolution model where without any external selective
pressure from treatment, acquisition of new genetic abnormalities
confer a proliferative advantage to a subclone at expense of others;
40% of patients progressed following a static progression model, where
all subclones were equally represented in both SMM and MM samples,
without any significant change in their subclonal structure.[34]
Gene Mutations in SMM
The
iStopMM study, a nationwide screening study for multiple myeloma
precursors in which all residents of Iceland 40 years or older are
involved, showed a prevalence of SMM in the total population of 0.53%
(0.67% in men and 0.39% in women); its prevalence increased in both
sexes with age.[35] In 193 individuals with SMM, the mean M-protein concentration was 0.62g/dL, and the median age was 70 years.[35]
Several
studies have explored the genetic alterations observed in SMM and the
genetic changes that underline its transition to MM. Using whole genome
sequencing, Bolli et al. showed that the genomic landscape, including
mutational profile and structural rearrangements at the SMM stage, is
very similar to that observed in MM.[34] Paired
sample analysis showed two patterns of progression: a static model,
implying the maintenance of the subclonal architecture during SMM
progression to MM, and the progression being related to the progressive
achievement of a sufficient disease burden; a spontaneous evolution
model implying changes at the level of subclonal composition.[31] The analysis of mutational signatures suggested a major role of APOBEC cytosine deaminases in disease progression.[34]
It
was estimated that patients with SMM have a higher risk of progression
to MM (10%/year) compared to those with MGUS (1%/year).[36]
Prognostic
models are unable to fully capture the risk of SMM progression since
also some patients evaluated as intermediate- or low-risk can still
progress to MM. The study of genomic profiles may help to define better
the risk of progression in SMM patients. Thus, Bustoros et al., through
whole genome sequencing of 214 patients with SMM, identified some
genetic predictors of SMM progression: thus, alterations of the MAPK
pathway (KRAS and NRAS mutations), the DNA repair pathway (deletion
p17, TP53, and ATM mutations) and MYC (translocations and copy number
alterations) are independent risk factors of progression after
accounting for clinical risk staging.[37]
Boyle
et al. reported the results of a study involving the analysis of 82
patients with SMM by targeted sequencing and comparing these results
with those observed in newly diagnosed MM and showed a lower frequency
of driver gene mutations in SMM compared to MM, a lower frequency on
NRAS and FAM46C mutations and fewer adverse translocations, del(1p),
del(14q), del(16q) and del(17p) in SMM than in MM, suggesting a
possible role of these genetic alterations as drivers of the transition
to MM; biallelic inactivation of tumor suppressor genes is markedly
less frequent in SMM; mutations in KRAS are associated with a shorter
time to progression.[38] The analysis of clonal
heterogeneity showed that changes in subclonal architecture precede
progression, and clonal diversity is a marker of time to progression.[38]
Bustoros
et al. reported the results of an integrative genetic analysis on 214
SMM patients using an unsupervised binary matrix factorization
clustering approach to identify molecular subtypes. Using this
approach, they identified six distinct genetic subtypes of SMM (Figure 2).[39]
 |
- Figure 2. Molecular
classification of SMM into six different clusters associated with
different molecular abnormalities, according to Bustoros et al.
|
A
hyperdiploid genotype characterizes cluster 1 (hyperdiploid-like 1) and
is significantly enriched in NRAS, TRAF3, and MAX mutations. Cluster 2
(hyperdiploid-like 2) is characterized by a high frequency of
hyperdiploidy (69%), frequent arm-level deletions, including 16q, 6q,
1p, 17p, 4q, 18q, and 20q and the IgH translocations t(14;20) and
enrichment in mutations of NRAS, BRAF, TP53, ATM, MAFB and CDKN2C
genes. Cluster 3 (translocation-like 1) is enriched in hypodiploid
tumors (<45 chromosomes) and is characterized by the presence of
t(4;14) which upregulates FGFR3 and MMSET genes, copy number losses of
14q, 1p, 8p, 10p, 11q, 12p, and 17p and by mutations in DIS3, MAF,
TGFR3, PRKD2, PRDM1 and HIST1H1E. Cluster 4 (hyperdiploid-like 3) is
characterized by the presence of hyperdiploid tumors that harbor
mutations in KRAS and NFKB1A genes and by MYC translocations. Cluster 5
(translocation-like 2) is characterized by overexpression of CCND1,
ERBB4, E2F7, E2F1, TRAK2, RBL1 and downregulation of DUSP4, TRAF6,
PRKD3, CCDC6 and ZNF844. Cluster 6 (hyperdiploid-like 4) is
characterized by hyperdiploidy, is enriched in NFKB2 and KLHL6
mutations, and copy gains in 2p.[39] Clusters 2, 3, and 4 are associated with an increased risk of progression to active MM (Figure 3).[39]
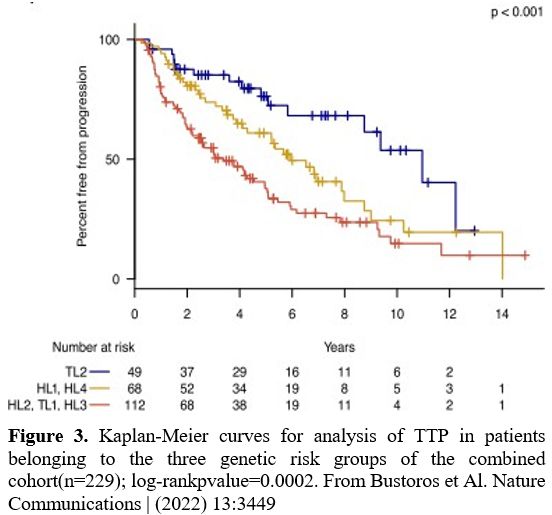 |
- Figure 3. Kaplan-Meier
curves for analysis of TTP in patients belonging to the three genetic
risk groups of the combined cohort(n=229); log-rankpvalue=0.0002. From
Bustoros et Al. Nature Communications | (2022) 13:3449
|
Patients
developing MM post-SMM (P-SMM) during clinical surveillance were
presenting with a lower disease burden, reduced level bone disease, and
potentially irreversible myeloma-defining events.[40]
Various
clinical risk models have attempted to evaluate the risk of SMM
progression. The Mayo risk evaluation criteria stratified SMM patients
into risk categories depending on no risk factors (low-risk), one risk
factor (low-risk), and two or more risk factors (high-risk); risk
factors include free light chain ratio >20, M-protein concentration
>2g/dL, BMPC percentage >20%.[41] This risk
evaluation system was updated by the International Myeloma Working
Group (IMWG), including some cytogenetic markers [t(4;14), t(14;16),
gain(1q) and del(13/13q)].[42] More recently, the
PANGEA model, based on the evaluation of M-protein levels, free light
chain ratio, age, creatinine concentration, BMPC percentage, and
hemoglobin trajectories, improved the prediction of SMM progression
compared with the two other models.[43] Other models of SMM stratification have been proposed, but there is significant discordance between them.[44]
Interestingly,
Diamond and coworkers have performed a whole genomic sequencing
analysis on 27 high-risk SMM (HR-SMM) patients treated with
carfilzomib, lenalidomide, and dexamethasone; after a median follow-up
of 52.8 months, median PFS was not reached and 51.9% of patients
achieved sustained MRD negativity.[45] The genomic
features of these patients were similar to those of ND-MM for that
concerns the frequency of t(4;14), t(14;16), and t(14;20); mutations of
NRAS were lower in HR-SMM than in ND-MM, as well as gene abnormalities
at MYC locus and gains of 1q; furthermore, aberrations of tumor
suppressor genes, such as CDKN2C, CYLD, TENT5C, FUBP1, MAX, NCOR1, NF1,
NFKBIA, PRMD1, RB1, RPL5 and TRAF3 were less frequent in HR-SMM than in
ND-MM.[42] Interestingly, the genomic features were
correlated with the treatment outcomes: gain 1q, t(4;14) and MYC
dysregulation through loss of MAX were associated with failure to
achieve MRD negativity; inactivation of CYLD, BREBBP, MAX, and t(4;14),
APOBEC expression, and chromotripsis all were associated with HR-SMM
progression; presence of any or more than one of these features was
associated with progression.[45]
SMM is
considered a heterogeneous disease entity which includes patients with
consistently variable risk of progression to MM; thus, in a subset of
patients, the disease is comparable to MGUS and exhibits a low rate of
MM progression, while in another subset of patients, is considered as
an early MM, with progression to symptomatic MM within 2 years. The
Mayo 2018 20/2/20 system classifies SMM patients into three subgroups,
low-, intermediate- and high-risk, based on the presence of 0, 1 or 2
or >2 risk factors, respectively, including >20% bone marrow
plasma cells, monoclonal protein >2g/dL, and free light chain ratio
>20.[41] The 2020 International Myeloma Working
Group risk stratification model further widened the separation of SMM
patients into four subgroups incorporating cytogenetic abnormalities
into the Mayo Clinic 2018 model.[42] The approach to
high-risk SMM patients varies among clinicians; while some advocate
early interventions, others reserve treatment at progression to MM. A
recent survey of 146 different clinicians showed that 92% of them did
not recommend routine treatment for high-risk SMM patients based on a
single time point assessment, instead preferring active surveillance.[46]
The active and frequent surveillance of these patients is important
because it was recently estimated that about 70% of HR-SMM patients
progress to MM in a follow-up of 3.9 years.[47]
A recent study strongly supports the important role of longitudinal evaluation of the evolution of risk biomarkers over time.[48]
Aklhagi et al. retrospectively evaluated the prognostic impact of risk
stratification in 398 SMM patients, who were analyzed at the Memorial
Sloan Kettering Cancer Center. They observed that risk stratification
based on the evaluation of biomarkers reflecting disease burden at the
time of diagnosis was unable to predict tumor progression in about 50%
of SMM patients who progressed to MM during the first year.[45]
In fact, among these rapidly progressing patients, only 43% had a
baseline M-protein ≥2.2 g/dL, and 43% had an FLCr ≥26; furthermore,
among these progressor patients, 29% had a baseline M-protein <1.6
g/dL and 26% had baseline FLCr <11.3.[48] However, the evolution of
these two biomarkers over time was predictive of risk of progression to
MM; thus, evolving changes in M-protein and FLCr were associated with a
higher risk of progression from SMM to MM: for patients with low-risk
baseline stratification, the presence of evolving M-protein (≥0.3 g/dL
increase) and eFLCr (≥50% increase), had a median time to progression
of 25 months, similar to that observed in patients with a baseline
high-risk.[48]
Abdallah et al. have reported the analysis of the mode of progression in 406 SMM patients evaluated at the Mayo Clinic.[49]
With a median follow-up of 3.9 years, 72% of the high-risk SMM patients
who did not receive treatment in the SMM phase progressed to MM; 11% of
the high-risk patients who received treatment at the SMM stage
progressed to MM.[49] The median time to progression
in the high-risk SMM patients was 2.6 years, compared to 7.0 years in
the non-high-risk patients.[49] Finally, a high
proportion (45%) of patients with high-risk SMM on active surveillance
develop end-organ damage at progression.[49]
Two
different strategies have been proposed for the treatment of HR-SMM:
either low-intensity regimens, such as lenalidomide and dexamethasone,
or intensive regimens with the aim of cure.[46] The
ensemble of the studies carried out until now do not support the early
intervention with intensive treatment strategies in SMM as the optimal
path to curing myeloma.[50] Interestingly, the
Immuno-PRISM trial evaluated the safety and the efficacy of
Teclistamab, a bispecific anti-CD38, and anti-CD3 monoclonal antibody,
in comparison to lenalidomide and dexamethasone for the treatment of
high-risk SMM patients.[51] In the Teclistamab
cohort, a 100% overall response (with 87% of CR and 13% of very good
partial responses) rate was observed, compared to 66% in the control
arm treated with lenalidomide and dexamethasone.[51] 100% of the patients treated with Teclistamab achieved an MRD-negative status.[51]
It is of interest to note that the ORR observed in high-risk SMM
patients was higher than that previously observed from R/ MM patients
treated with Teclistamab (100% vs 63%, respectively).
Conclusions
The
development of new techniques for the analysis of genomic alterations
occurring in MM and its precursors, MGUS and SMM, have greatly
contributed to defining the acquired genomic abnormalities involved in
tumor initiation and progression. MGUS and SMM are heterogeneous
conditions with the presence of tumors with distinct pathogenic
phenotypes and clinical outcomes. The identification of SMM patients
with a molecularly defined high risk of progression to MM offers the
unique opportunity of early intervention with a therapeutic approach on
a low tumor burden using drugs such as bispecific antibodies with a
good safety profile.
References
- Huang J, Chan SC, Lok V, Zhang L, Lucero-Prisno DE 3rd, Xu
W, Zheng ZJ, Elcarte E, Withers M, Wong MCS; Non-communicable Disease
Global Health Research Group, Association of Pacific Rim Universities.
The epidemiological landscape of multiple myeloma: a global cancer
registry estimate of disease burden, risk factors, and temporal trends.
Lancet Haematol. 2022 Sep;9(9):e670-e677. doi:
10.1016/S2352-3026(22)00165-X.
https://doi.org/10.1016/S2352-3026(22)00165-X PMid:35843248
- Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S,
Cerhan JR, Rajkumar V. Long-term follow-up of monoclonal gammopathy of
undermined significance. N Engl J Med 2018; 378: 241-249.
https://doi.org/10.1056/NEJMoa1709974 PMid:29342381 PMCid:PMC5852672
- Murray D, Kumar SK, Kyle RA, Dispenzieri A, Dasari S,
Larson DR, Vachon C, Cerhan JR, Rajkumar SV. Detection and prevalence
of monoclonal gammopathy of undetermined significance: a study
utilizing mass spectrometry-based monoclonal immunoglobulin rapid,
accurate mass measurement. Blood Cancer J 2019; 9: 102.
https://doi.org/10.1038/s41408-019-0263-z PMid:31836698
PMCid:PMC6910906
- Vachon C, Murray J, Allmer C, Larson D, Norman AD, Sinnwell
JP, Disopenzieri A, Kleinstern G, Visram A, Kyle RA, et al. Prevalence
of heavy chain MGUS by race and family history risk groups using a
high-sensitivity screening method. Blood Adv 2022; 6: 3746-3750.
https://doi.org/10.1182/bloodadvances.2021006201 PMid:35316833
PMCid:PMC9631569
- El-Khoury H, Lee DJ, Alberge JB, Redd R, Cea-Curry CJ,
Perry J, Barr H, Murphy C, Sakrikar D, Barnidge D, et al. Prevalence of
monoclonal gammopathies and clinical outcomes in a high-risk US
population screened by mass spectrometry: a multicentre cohort study.
Lancet Hematol 2022; 9: e340-e349.
https://doi.org/10.1016/S2352-3026(22)00069-2 PMid:35344689
- Lee DJ, El-Khoury H, Tramontano AC, Alberge JB, Perry J,
Davis MI, Horowitz E, Redd R, Sakrikar D, Barnidge D, et al. Mass
spectrometry MGUS is associated with obesity and other modifiable risk
factors in a high-risk population. Blood Adv 2024; in press.
https://doi.org/10.1182/bloodadvances.2023010843 PMid:38212245
PMCid:PMC10997907
- Boddicker NJ, Parikh SA, Norman AD, Rabe KG, Griffin R,
Gall TG, Robinson DP, Olson JE, Dispenzieri A, Rajkumar V, et al.
Relationship among three common hematological premalignant conditions.
Leukemia 2023; 37: 1719-1722.
https://doi.org/10.1038/s41375-023-01914-z PMid:37147423
PMCid:PMC10400408
- Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B,
Shaughnessy J. Cyclin D regulation: an early and unifying pathogenic
event in multiple myeloma. Blood 2005; 106: 296-303.
https://doi.org/10.1182/blood-2005-01-0034 PMid:15755896
PMCid:PMC1895118
- Morgan GJ, Walker BA, Davies FE. The genetic architecture
of multiple myeloma. Nat Rev Cancer 2012; 12: 335-348.
https://doi.org/10.1038/nrc3257 PMid:22495321
- Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple
myeloma and its premalignant precursor. J Clin Invest 2012; 122:
3456-3463. https://doi.org/10.1172/JCI61188 PMid:23023717
PMCid:PMC3461901
- Samur AA, Minelli S, Shammas M, Falciniti M, Magrangeas F,
Richardson PG, Moreau P, Attal M, Anderson KC, Parmigiani G, et al.
Deciphering the chronology of copy number alterations in multiple
myeloma. Blood Cancer 2019; 9: 39.
https://doi.org/10.1038/s41408-019-0199-3 PMid:30914633
PMCid:PMC6435669
- Zhao S, Choi M, Henck C, Mane S, Barlogie B, Lifton RP,
Dodapakar MV. Serial exome analysis of disease progression in
premalignant gammopathies. Leukemia 2014; 28: 1548-1552.
https://doi.org/10.1038/leu.2014.59 PMid:24496302 PMCid:PMC4142199
- Mikulasova A, Smetana J, Wayhelova M, Janyskova H, Sandecka
V, Kufova Z, Almasi M, Jarkovsky J, Gregora E, Kessler P, et al.
Genome-wide profiling of copy-number alteration in monoclonal
gammopathy of undetermined significance. Eur J Haematol 2016; 97:
568-575. https://doi.org/10.1111/ejh.12774 PMid:27157252
- Marchesini M, Ogoti Y, Fiorini E, Samur AA, Nexi L, D'Anca
M, Storti P, Samur MK, Ganan-Gomez I, Fulciniti MT, et al. ILF2 is a
regulator of RNA splicing and DNA damage response in 1q21-amplified
multiple myeloma. Cancer Cell 2017; 32: 88-100. The prognostic role of
1q21 gain/amplification in newly diagnosed multiple myeloma: the
faster, the worse. Cancer 2023; 129: 1005-1016.
https://doi.org/10.1002/cncr.34641 PMid:36704927
- Razuma Y, Shirakawa K, Tashiro Y, Yamazaki H, Nomura R,
Horisawa Y, Takeuchi S, Stanford E, Konishi Y, Matsui H, et al. ILF2
enhances the DNA cytosine deaminase activity of tumor mutator APOBEC3B
in multiple myeloma cells. Scient Rep 2022; 12:
2278. https://doi.org/10.1038/s41598-022-06226-3 PMid:35145187
PMCid:PMC8831623
- Wang Y, Xu J, Xu B, Li P, Yang Y, Wang W, Xu T, Maihemaiti
A, Lan T, Wang P, et al. The prognostic role of 1q21 gain/amplification
in newly diagnosed multiple myeloma: the faster, the worse. Cancer
2023; 2023; 129: 1005-1016. https://doi.org/10.1002/cncr.34641
PMid:36704927
- Boyd KD, Ross FM, Walker BA, Wardell CP, Tapper WJ,
Chiecchio L. Mapping of chromosome 1p deletions in myeloma identifies
FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions
associated with adverse survival. Clin Cancer Res 2011; 17:7776-7784.
https://doi.org/10.1158/1078-0432.CCR-11-1791 PMid:21994415
PMCid:PMC5751883
- Leone PE, Walker BA, Jenner MW, Chiecchio L, Dagrada GP,
Protheroe RK. Deletions of CDKN2C in multiple myeloma: biological and
clinical implications. Clin Cancer Res 2008; 16: 6033-6041.
https://doi.org/10.1158/1078-0432.CCR-08-0347 PMid:18829482
PMCid:PMC2581792
- Chiecchio L, Dagrada GP, Ibrahim AH, Dachs Cabanas E,
Protheroe RK. Timing of acquisition of deletion 13 in plasma cell
dyscrasias is dependent on genetic context. Haematologica 2009; 94:
1708-1713. https://doi.org/10.3324/haematol.2009.011064 PMid:19996118
PMCid:PMC2791926
- Mikulasova A, Wardell CP, Murison A, Boyle EM, Jackson GH,
Smetana J, Kufova Z, Pour L, Sandecka V, Almasi M, et al. The spectrum
of somatic mutations in monoclonal gammopathy of undetermined
significance indicates a less complex genomic landscape than that in
multiple myeloma. Haematologica 2017; 102: 1617-1625.
https://doi.org/10.3324/haematol.2017.163766 PMid:28550183
PMCid:PMC5685224
- Landgren O. Advances in MGUS diagnosis, risk
stratification, and management: introducing myeloma-defining genomic
events. Am Soc Hematol Educational Program 2021; 2021: 662-672.
https://doi.org/10.1182/hematology.2021000303 PMid:34889381
PMCid:PMC8791104
- Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Adbasal
F, Zheng-Lin B, Yellapantula V, Derkach A, Geerdens E, Diamond BT, et
al. Whole-genome sequencing reveals progressive versus stable myeloma
precursor conditions as two distinct entities. Nat Commun 2021; 12:
1861. https://doi.org/10.1038/s41467-021-22140-0 PMid:33767199
PMCid:PMC7994386
- Varrettoni M, Zibellini S, Defrancesco I, Ferretti VV,
Rizzo E, Malcovati L, Galli A, Della Porta MG, Boveri E, Arcaini L, et
al. Pattern of somatic mutations in patients with Waldenstrom
macroglobulinemia or IgM monoclonal gammopathy of undetermined
significance. Haematologica 2017; 102: 2077-2083.
https://doi.org/10.3324/haematol.2017.172718 PMid:28983055
PMCid:PMC5709107
- Varrettoni M, Zibellini S, Boveri E, Klersy C, Candido C,
Rattotti S, Ferretti VV, Defrancesco I, Mangiacavalli S, Nizzoli E, et
al. A risk stratification model on the initial concentration of the
serum monoclonal protein and MYD88 mutation status identifies a subset
of patients with IgM monoclonal gammopathy of undetermined significance
at high risk of progression to Waldenstrom macroglobulinemia or other
lymphoproliferative disorders. Brit J Haematol 2019; 187: 441-446.
https://doi.org/10.1111/bjh.16086 PMid:31276195
- Khwaja J, D'Sa S, Minnema MC, Kersten MJ, Wechalekar A, Vos
J. IgM monoclonal gammopathies of clinical significance: diagnosis and
management. Haematologica 2022; 107: 2037-2046.
https://doi.org/10.3324/haematol.2022.280953 PMid:35770530
PMCid:PMC9425303
- Cao XX, Meng Q, Cai H, He TH, Zhang CL, Su W, Sun J, Li Y,
Xu W, Zhou DB, Li J. Detection of MYD88 L265P and WHIM-like CXCR4
mutation in patients with IgM monoclonal gammopathy related disease.
Ann Hematol 2017; 96: 971-976.
https://doi.org/10.1007/s00277-017-2968-z PMid:28280994
- Moreno DF, Lopez-Guerra M, Paz S, Oliver-Caldés A, Mena MP,
Correa JG, Battram AM, Osuna M, Rivas-Delgado A, Rodriguez-Lobato LG,
et al. Prognostic impact of MYD88 and CXCR4 mutations assessed by
droplet digital polymerase chain reaction in IgM monoclonal gammopathy
of undetermined significance and smouldering Waldenstrom
macroglobulinemia. Brit J Haematol 2023; 200: 187-196.
https://doi.org/10.1111/bjh.18502 PMid:36210485 PMCid:PMC10092069
- Stern S, Chaudhuri S, Drayson M, Henshaw S, Karunanithi S,
Willis F. Investigation and management of the monoclonal gammopathy of
undetermined significance. Brit j Haematol 2023; 202: 734-744.
https://doi.org/10.1111/bjh.18866 PMid:37587091
- Rajkumar SV, Kyle RA, Therneau TM, Melton LJ, Bradwell AR,
Clkark RJ. Serum free light chain ratio is an independent risk factor
for progression in monoclonal gammopathy of undetermined significance.
Blood 2005; 106: 812-817. https://doi.org/10.1182/blood-2005-03-1038
PMid:15855274 PMCid:PMC1895159
- Bogun L, Koch A, Scherer B, et al. Stromal alterations in
patients with monoclonal gammopathy of undetermined significance,
smoldering myeloma and multiple myeloma. Blood Adv 2024; in press.
- Lopez-Corral L, Sarasquete ME, Bae S, Garcia-Sanz R, Mateos
MV, Corchete LA, Sayagués JM, Garcia EM, Bladé J, Oriol A, et al.
SNP-based mapping arrays reveal high genomic complexity in monoclonal
gammopathies, from MGUS to myeloma status. Leukemia 2012; 26:
2521-2529. https://doi.org/10.1038/leu.2012.128 PMid:22565645
- Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC,
Kaiser MF, Mirabella F, Lopez-Corral L, Humphray S, Murray L, et al.
Intraclonal heterogeneity is a critical early event in the development
of myeloma and precedes the development of clinical symptoms. Leukemia
2014; 28: 384-390. https://doi.org/10.1038/leu.2013.199 PMid:23817176
PMCid:PMC3916874
- Dutta AK, Fink JL, Grady JP, Morgan GJ, Mullighan CG, To
LB, Hewett DR, Zannettino A. Subclonal evolution in disease progression
from MGUS/SMM to multiple myeloma is characterized by clonal stability.
Leukemia 2019; 33: 457-468. https://doi.org/10.1038/s41375-018-0206-x
PMid:30046162 PMCid:PMC6365384
- Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam
A, Martincorena I, Dawson KJ, Samur MK, Zamora J, et al. Genomic
patterns of progression in smoldering multiple myeloma. Nat Commun
2018; 9. 3363. https://doi.org/10.1038/s41467-018-05058-y PMid:30135448
PMCid:PMC6105687
- ThorsteinsdottirS; Gislason SK, Aspelund T, Rognvaldsson S,
Oskarson JP, Siguroardottir AR, Vioarsson B, Onundarson PT, Agnarsson
BA, Siguroardottir M, etal. Prevalence of smoldering multiple myeloma
based on nationwide screening. Nature Medi 2023; 29: 467-472.
https://doi.org/10.1038/s41591-022-02183-6 PMid:36747117
- Kyle RA, Ranistein ED, Therneau ED. Clinical course and
prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med
2017; 356: 2582-2590. https://doi.org/10.1056/NEJMoa070389
PMid:17582068
- Bustoros M, Sklavenitis-Pitsofidis R, Park J, Redd R,
Zhitomirksky B, Dunford AJ, Salem K, Tai YT, Anand S, Mouhieddine TH,
et al. Genomic profiling of smoldering multiple myeloma identifies
patients at a high risk of disease progression. J Clin oncol 2020; 38:
2380-2389. https://doi.org/10.1200/JCO.20.00437 PMid:32442065
PMCid:PMC7367550
- Boyle EM, Deshpande S, Tytarenko R, Ashby C, Wang Y, Bauer
MA, Johnson SK, Wardell COP, Thanendrajan S, Zangari M, et al. The
molecular makeup of smoldering myeloma highlights the evolutionary
pathways leading to multiple myeloma. Nat Commun 2021; 12: 293.
https://doi.org/10.1038/s41467-020-20524-2 PMid:33436579
PMCid:PMC7804406
- Bustoros M, Anand S, Sklavenitis-Pistofidis R, Redd R,
Boyle EM, Zhitomirsky B, Dunford AJ, Tai YT, Chavda SJ, Boehner C, et
al. Genetic subtypes of smoldering multiple myeloma are associated with
distinct pathogenic phenotypes and clinical outcomes. Nat Commun 2022;
13: 3449. https://doi.org/10.1038/s41467-022-30694-w PMid:35705541
PMCid:PMC9200804
- Cohen MM, Fridberg G, Cohen I, Robinson R, Vaxman I,
Shragai T, Trestman S, Ziv-Baran T, Melamed N, Raanani P, et al.
Smoldering multiple myeloma progressing to active multiple myeloma vs
de-novo active multiple myeloma - a comparison of disease
characteristics, organ involvement and outcomes in a real-life
multicenter retrospective cohort study. Blood 2023; 142 (suppl. 1):
4728. https://doi.org/10.1182/blood-2023-186140
- Lakshman A, Rajkumar SV, Buadi FK, Binder M, Gertz MA, Lacy
MQ, Dispenzieri A, Dingli D, Fonder AL, Haymann SR, et al. Risk
stratification of smoldering multiple myeloma incorporating revised
IMWG diagnostic criteria. Blood Cancer J 2018; 8: 59.
https://doi.org/10.1038/s41408-018-0077-4 PMid:29895887
PMCid:PMC5997745
- Matoes MV, Kumar S, Dismopoulos MA, Gonzalez-Calle V,
Kastritis E, Hajek R, De Larrea CF, Morgan GJ, Merlini G, Golschmidt H,
et al. International Myeloma Working Group risk stratification model
for smoldering multiple myeloma (SMM). Blood Cancer J 2020; 10: 102.
https://doi.org/10.1038/s41408-020-00366-3 PMid:33067414
PMCid:PMC7567803
- Cowan A, Ferrari F, Freeman SS, Redd R, El-Khoury H, Perry
J, Patel V, Kaur P, Barr H, Lee DJ, et al. Personalised progression
prediction in patients with monoclonal gammopathy of undetermined
significance for smouldering multiple myeloma (PANGEA): a
retrospective, multicohort study. Lancet Hematol 2023; 10: e203-212.
https://doi.org/10.1016/S2352-3026(22)00386-6 PMid:36858677
- Hill E, Dew A, Morrison C, Yuan C, Stetler-Stevenson M,
Landgren O, Kazandjian D. Assessment of discordance among smoldering
multiple myeloma risk models. JAMA Oncol 2021; 7: 132-134.
https://doi.org/10.1001/jamaoncol.2020.5585 PMid:33211080
PMCid:PMC7677870
- Diamond B, Kazandjian D, Papadimitriou M, Ziccheddu B,
Blaney P, Chojnacka M, Durante M, Hill E, Sklavenitis-Pistofidis R,
Maclachlan KH, et al. Genomic profiling to interpret the outcomes of
early intervention for high-risk smoldering myeloma. Blood 2023; 142
(suppl.1): 757. https://doi.org/10.1182/blood-2023-181817
- Mohyuddin GR, Chakraborty R, Cliff ES, Derman BA. Clinician
preferences on treatment of smoldering myeloma: a cross-sectional
survey. eClinical Medicine 2023; 65: 102272.
https://doi.org/10.1016/j.eclinm.2023.102272 PMid:38046471
PMCid:PMC10689285
- Rajkumar S, Abdallah N, Lakshman A, Kumar S, Cook J, Binder
M, Kapoor P, Dispenzieri A, Gertz M, Lacy M, et al. Mode of progression
in smoldering multiple myeloma: a study of 406 patients. Res Sq 2023;
23:
rs.3.rs-337834. https://doi.org/10.21203/rs.3.rs-3378634/v1
- Akhlagi T, Nemirovsky D, Maclachlan KH, Korde N, Mailankody
S, Lesokhin A; Hassoun H, Patel D, Shah UA, Tan C, et al. Evaluating
the effect of involving changes in serum biomarkers on the risk of
progression in smoldering multiple myeloma. Blood 2023; 142 (suppl.1):
877. https://doi.org/10.1182/blood-2023-180315
- Abdallah NH, Lakshman A, Kumar SK, et al. Mode of
progression in smoldering multiple myeloma: a study of 406 patients.
Blood Cancer J 2024 Jan; 4: 9.
https://doi.org/10.1038/s41408-024-00980-5 PMid:38228628
PMCid:PMC10791688
- Chakraborty R, Al Hadidi S, Cliff ERS, Mohyuddin GR. Is
aggressive treatment of smoldering myeloma the path to curing myeloma?
Blood Adv 2023; 7: 3932-3934.
https://doi.org/10.1182/bloodadvances.2023009658 PMid:37196639
PMCid:PMC10405196
- Nadeem O, Magidson S, Midha S, O'0Donnell E, Hartley-Brown
MA, Seprling AS, Redd RA, Marto M, Davie C, Ricciardi C, et al.
Immuno-PRISM: a randomized phase II platform study of bispecific
antibodies in high-risk smoldering myeloma. Blood 2023; 142 (suppl.1):
206. https://doi.org/10.1182/blood-2023-177954