In March 2004, a 49-year-old man was diagnosed with AML M4 with the inversion of chromosome 16. He started induction chemotherapy with daunorubicine, cytarabine and thioguanine, achieving complete remission (CR). He then underwent 3 consolidation courses of chemotherapy with high-dose Cytosine. In January 2006, he developed relapsed disease and started salvage chemotherapy with FLAG-Ida [fludarabine, cytarabine, G-CSF (granulocyte-colony stimulating factor), idarubicin], to which he responded well and went into complete remission; he subsequently received a second cycle of FLAG-Ida as consolidation. He underwent a matched volunteer unrelated haematopoietic stem cell transplant (10/10 HLA match) with Fludarabine 150 mg/msq, Busulphan 6.4 mg/Kg and Alemtuzumab (Campath) 60 mg (FBC) on 29th June 2006. Graft-versus-host disease (GVHD) prophylaxis consists of a Cyclosporine single agent. On D+76, while cyclosporine was being tapered, he developed acute grade 2 skin GVHD, which progressed to chronic GVHD requiring treatment with Psoralen plus UltraViolet A radiation (PUVA) and prolonged systemic immune suppression. Cyclosporine was stopped one-year post-transplant. Repeated BM examinations, until five years post-transplant confirmed molecular remission (CBFb/MYH11 by Real-Time PCR). In 2016, routine evaluation revealed neutropenia and reflex testing with a bone marrow showed molecular Minimal Residual Disease (MRD) negative AML, but granulocytic hypoplasia and the presence of a small non-clonal T-LGL expansion (4% T cells with CD3+, CD57+, CD5+, CD16neg, CD56neg, CD2+, CD7+ and alpha/beta +). He was initiated on prednisolone and G-CSF, to which there was an improvement. However, on tapering of steroids, the cytopenia recurred, necessitating the use of cyclosporin followed by Sirolimus as a steroid-sparing agent. The blood counts normalised until December 2022, when he again developed profound neutropenia. A repeat bone marrow examination revealed para-trabecular and interstitial infiltrate of B cells, likely monomorphic PTLD consistent with Diffuse Large B Cell Lymphoma (DLBCL) of non-GC type [PET CT did not reveal any nodal disease, and EBV viral load showed a peak of 25000 DNA copies/ml]. Treatment with Rituximab 375mg/m2 weekly for four weeks was commenced, and that resulted in complete resolution of cytopenia, and the end of treatment bone marrow confirmed in February 2023 the absence of AML (MRD negative by flowcytometry and RTPCR) and DLBCL. Due to the persistence of an enlarged LGL population (7% T LGL cells), he was continued on prednisolone 5mg and Sirolimus 2 mg. In September 2023, he had a new episode of pancytopenia, and bone marrow showed relapsed high-grade B-cell lymphoma (very low volume, ~1-2%) with AML molecular remission. PET CT revealed generalised lymphadenopathy on both sides of the diaphragm. A Lymph node biopsy revealed EBV-positive DLBCL, and he has been commenced on R-CHOP chemotherapy.
A biphasic pattern of PTLD has been recognised, with early disease within the first year of transplant and a second peak 7 to 10 years after transplant.[3]
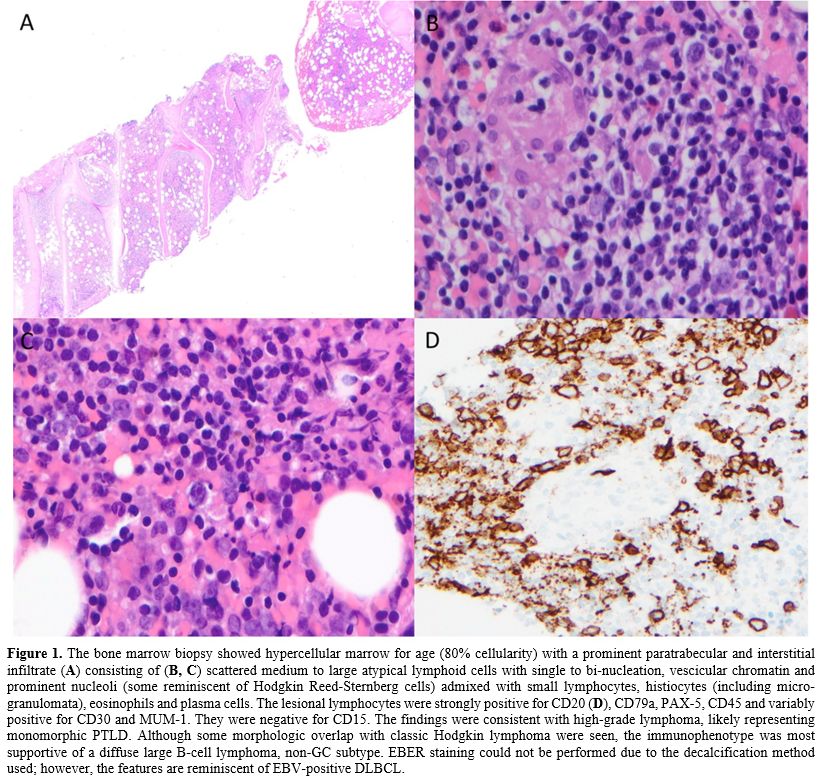 |
|
Presentations after ten years of transplant have been defined as extremely late PTLD, and this is the first report of an adult presenting with it in the post-HCT setting, though there are few reports in the paediatric and post-SOT setting.[2]
For HCT, PTLD risk factors include EBV recipient-donor sero-mismatch, selective donor T-cell depletion, haploidentical donor, unrelated or HLA-mismatched related donor, umbilical cord blood transplant, use of reduced intensity conditioning regimens, age of recipient older than 50, the use of anti-thymocyte globulin (ATG) or anti-CD3 monoclonal antibody and chronic GvHD requiring prolonged immunosuppression.[5] Much of the data for PTLD, and especially very late PTLD, comes from solid organ transplants.[6] PTLD is lowest after HCT and liver transplants compared to the other transplants[7] since immune suppression is typically stopped post-HCT if there are no complications like chronic GVHD. In the landmark large series describing 18,014 patients who underwent allogeneic bone marrow transplantation (BMT) at 235 centres worldwide,[7] the authors concluded that altered immunity and T-cell regulatory mechanisms like chronic GVHD were responsible for PTLD. Notably, EBV and CMV reactivation did not correlate with late-onset PTLD.[8] The use of Campath and ATG has been implicated in the risk of developing PTLD.[3,9] In our case report, there was the presence of chronic GVHD necessitating the use of systemic immune suppression until 1-year post-transplant, followed by nearly six years of treatment of immune cytopenia with prednisolone, cyclosporin and Sirolimus prior to developing the PTLD. Thus, there is a need to recognise atypical risk factors for late-onset PTLDs and to keep the suspect of this entity in the differential diagnosis process.
In the absence of EBV as a driver, other postulated theories include the presence of lymphoid load in the allograft and chronic antigen stimulation, which may result in a dysregulated immune response leading to PTLD.[10] The major factors influencing both B cell and T cell immune reconstitution are GVHD and the use of immune suppressive agents. Generally, B cell numbers recover to normal counts within 12 months after HSCT,[11] although complete recovery may take up to 2 years. Patients receiving antithymocyte globulin-fresenius (ATG-F) are known to have delayed CD19+ B cell recovery compared to non-ATG patients,[12] and Campath is well known to offer a deeper lymphodepletion resulting in even later immune reconstitution.[13] T lymphopenia and inadequate repertoire of CD4+ and CD8+ T cells, lasting 1 year or more after transplant, foster recurrent infections with latent viruses.[14]
The optimal treatment of PTLD is still evolving. Quick withdrawal or reduction of immune (RIS) suppression and induction therapy with weekly rituximab for CD20-positive cases is the standard of care for PTLD.
The option of RIS alone may not be feasible because of the risk of GVHD[5] and because the still immune incompetent host may not be able to mount a cytotoxic response to halt the proliferative process.[6] However, the response rates to rituximab monotherapy in HSCT PTLD have been reported to be only 20% (ORR 60%–65%) with a 2-year median OS of 50%,[7] hence second line options practically include chemotherapy (R-CHOP), Immunotherapy with Brentuximab if CD30+ and ideally EBV specific Cytotoxic Lymphocyte therapy (CTLs) where available to offset infective complications and regimen related toxicities of systemic chemotherapy. This patient responded to first-line therapy despite the bone marrow involvement, which has been described to have worse outcomes.
In conclusion, this case highlights the complexity of very prolonged immune suppressive therapy and shows that very late-onset PTLS post-HCT can occur in a setting of persistent immunosuppression.