Multiple
myeloma (MM) is a disorder of the monoclonal plasma cells and is the
second most common hematologic malignancy. MM initiation and
progression are dependent upon complex genomic abnormalities. The
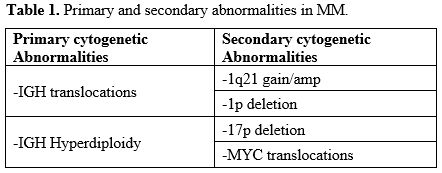
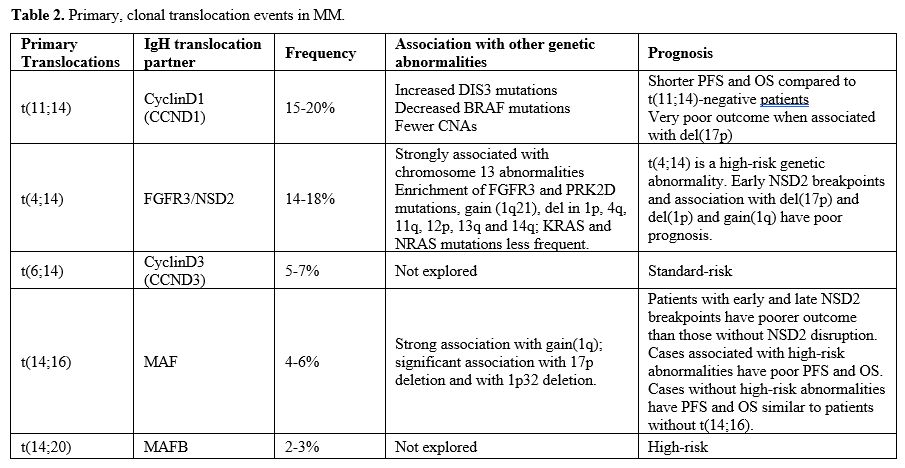
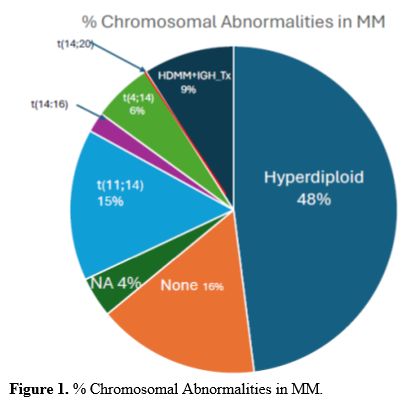
current pathogenic model of MM includes two types of primary events,
represented by chromosome translocations or chromosome number
alterations resulting in hyperdiploidy. These primary molecular events
are observed both in MM and in monoclonal gammopathy, its premalignant
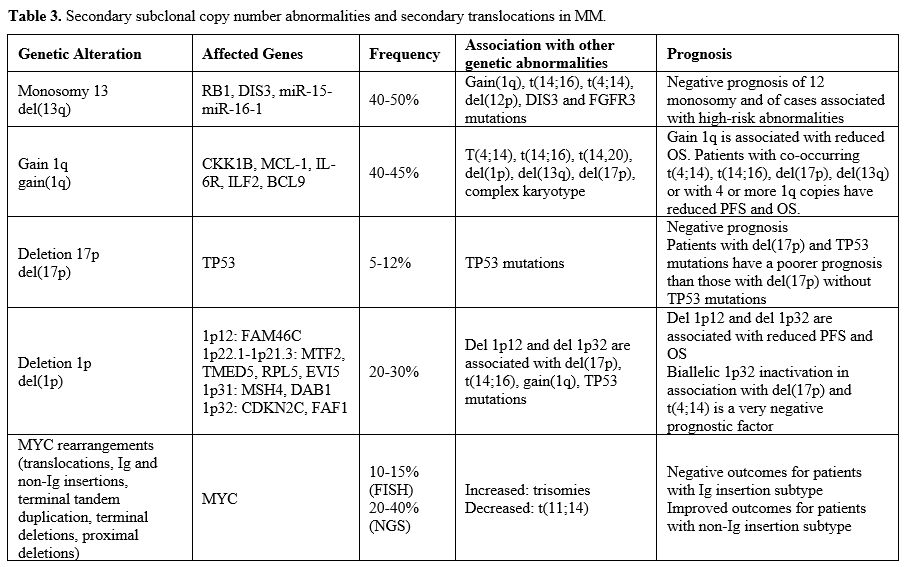
precursor. Subsequent genetic events allow the progression of
monoclonal gammopathy to MM and, together with primary events,
contribute to the genetic complexity and heterogeneity of MM.
Newer
therapies have considerably improved patient outcomes; however, MM
remains an incurable disease and most patients experience multiple
relapses.
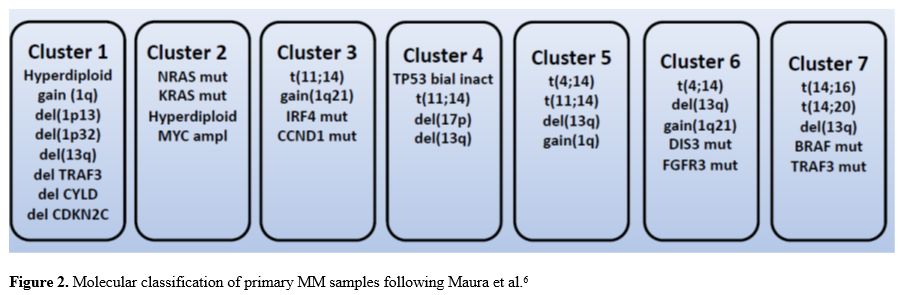
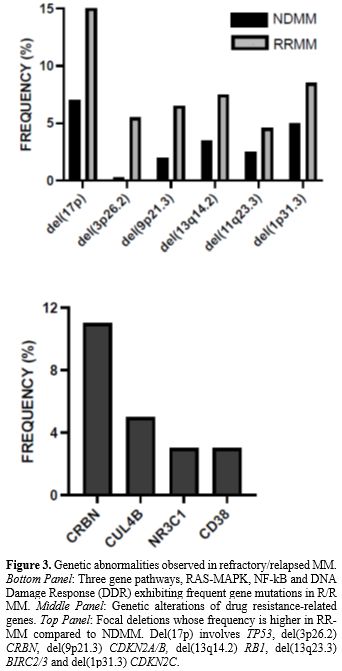
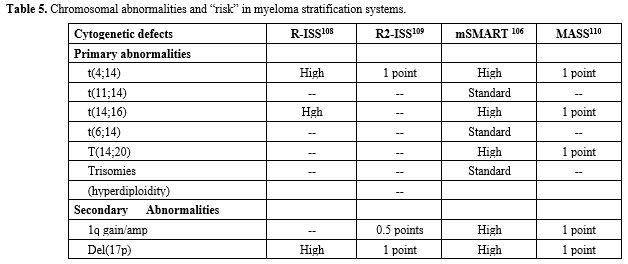
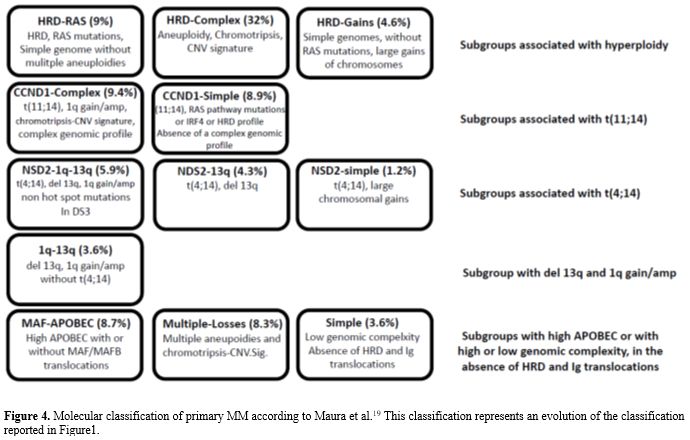
The dramatic progresses achieved in the analysis of the
heterogeneous molecular features of different MM patients allowed a
comprehensive molecular classification of MM and the definition of an
individualized prognostic model to predict an individual MM patient’s
response to different therapeutic options. Despite these progresses,
prognostic models fail to identify a significant proportion of patients
destined to early relapse. Treatment strategies are increasingly. Based
on disease biology, trials are enriched for high-risk MMs, whose
careful definition and categorization requires DNA sequencing studies.