 |
|
 |
|
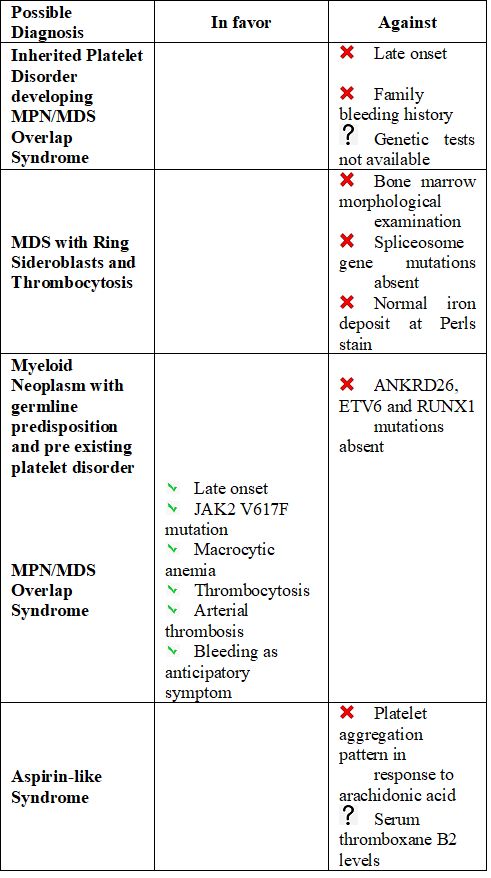
A possible MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) was excluded, as ring sideroblasts were not appreciable and spliceosome gene mutations were not detected.
The patient was treated with hydroxyurea (4.5 g/week), resulting in a stable decrease of platelet count below 600 x 109/L. Hemoglobin levels significantly increased with rEPO 30.000 UI/week. An abdomen ultrasound exam showed the stability of the spleen six months after treatment had been started. A platelet aggregation test was repeated, confirming the same abnormalities shown previously. Antiplatelet prophylaxis has not yet been started despite the myeloproliferative aspect of the neoplasm and ischemic encephalopathy.
Certainly, the late onset of bleeding diathesis and the absence of hematologic neoplasms in family members made the hypothesis of a hereditary disease quite unlikely. We did not support the possibility of platelet disorders with a de novo germline predisposition to myeloid neoplasms as our patient was RUNX1/ ANKRD26/ETV6 negative; moreover, in this case, we would have expected a thrombocytopenic rather than a patient with thrombocytosis.
The reduced platelet response to so-called "weak agonists" (epinephrine) could suggest an "aspirin-like syndrome"[4] despite normal platelet aggregation in response to arachidonic acid. Serum thromboxane B2 levels would have been useful,[5] but they were not available for this patient.
We assumed the more likely hypothesis that platelet dysfunction could be the epiphenomenon of the hematological neoplasm that would have been diagnosed later.
Little information is available about platelet function and its role in many diseases, including MPN.[6] However, despite more infrequent than thrombosis, bleeding in MPN represents a more common phenomenon than in the general population[7] attributable 1) to a qualitative defect of von Willebrand Factor,[8] also in patients with controlled platelet count;[9] 2) a reduced expression or function of glycoproteins (GpIb or Gp IIb-IIIa);[10] 3) abnormalities in platelet granules.[11] As a consequence, a defective platelet aggregation pattern may be present in MPN, except for arachidonic acid and ristocetin as agonists.[10]
In conclusion, in this case, the hemorragic phenotype preceded the oncohematological diagnosis, appearing as the epiphenomenomen of the most prominent Myeloproliferative features in the setting of an MDS/MPN Overlap Syndrome.
The long-term outcome of this patient will definitely clarify the hematological diagnosis and the relationship between bleeding and oncohematological diagnosis.