The b-like gene cluster contains embryonic (e) fetal (g1 and g2) and adult (b and d) globin genes, spatially ordered in their ontogenic time of expression during embryonic, fetal, and adult life. Particularly, the silencing of g fetal globin genes is dependent upon reduced stimulation by transcriptional activators and a repressive effect mediated by repressor transcriptional complexes, including transcription factors such as BCL11A and LRF.
Mutations in the adult b-globin gene cause b-thalassemia (b-thal) and sickle cell disease (SCD), two autosomal recessive diseases of red blood cells with worldwide diffusion. In b-thalassemia, more than 400 mutations in the b-globin gene cluster, including point mutations, minor deletions or insertions, or gross deletions in either the b-globin gene or in its flanking, non-coding regions, have been reported. These result in reduced production of b-globin and markedly decreased HbA synthesis, with an excess of free a-chains that are unstable, precipitate, and induce oxidative damage and premature death of erythroid cells.
SCD is caused by a point mutation of the—globin gene, which determines the production of an abnormal hemoglobin (HbS) in which the glutamic acid at position 6 is replaced by a Valine residue. This changes a negatively charged side chain into a hydrophobic one, allowing the polymerization of deoxygenated HbS. This alters RBC architecture and flexibility to the sickle cell shape.
In recent years, several trials have been developed to investigate the safety and efficacy of gene therapy approaches for the treatment of patients with transfusion-dependent b-thal and SCD. Basically, there are two different approaches to gene therapy for hemoglobinopathies based on the genetic engineering of autologous hematopoietic stem cells (HSCs): (i) gene addition strategies based on lentiviral vectors to add functional copies of the gene encoding b-globin in defective HSCs; (ii) gene editing strategies based on the use of CRISPR-Cas9, transcription activator-like effector protein nuclease and zinc finger nuclease techniques either to directly repair the underlying genetic cause of disease or to induce fetal hemoglobin production by gene disruption.
Initial studies were based on gene addition strategies and have largely supported the safety and clear efficacy of this gene therapy approach. Particularly, these studies have shown a good risk/benefit ratio, with no insertional mutagenesis and major adverse events related to myeloablative conditioning or to the underlying pathology.[1] These studies also showed that the inclusion in the lentiviral vectors of a surrogate version of the globin LCR limits the expression of the b-globin transgene generated from a vector copy number (VCN) per cell compatible with a safe profile.[2] In fact, in b-thal patients, to improve clinical outcome, it was required to optimize the engineering of HSCs through increasing VCN and transduction efficiency.[3]Gene editing strategies do not have this type of limitation.
Several recent trials have explored the safety profile and the effectiveness of gene editing strategies in b-thalassemia and SCD patients.
BCL11A, the Main Regulator of HbF Synthesis, is a
Key Target of Gene Editing for the Treatment of Hemoglobinopathies
HbF (a2g2) synthesis
level is genetically controlled and modifies the severity of
hemoglobinopathies, such as b-thal and SCD. BCL11A gene encodes a
zinc-finger protein predominantly expressed in brain and hematopoietic
tissue. BCL11A functions mainly as a transcriptional repressor that is
crucial in brain, hematopoietic system development, as well as
fetal-to-adult hemoglobin switching. The expression of this gene is
regulated by transcription factors, microRNAs, and genetic variations.
Particularly, the transcription factor BCL11A is a major regulator of
HbF synthesis during development. BCL11A acts as a transcriptional
repressor of g-globin gene expression through the occupation of
g-globin genes to inhibit their expression in adult erythroblasts.4
Particularly, a zinc finger cluster present in the BCL11A protein
interacts with a specific DNA sequence present in the promoters of g1
and g2 globin genes, repressing their transcriptional activity.[4]
BCL11A expression is transcriptionally controlled during development. In fetal erythroid cells, BCL11A expression is inhibited by the repressor HIC2, whose expression is high in fetal cells but low in adult erythroid cells.[5] In turn, HIC2 expression is controlled by some microRNAs pertaining to the let-7 miRNA family, which act as repressors of HIC2 expression.[6]
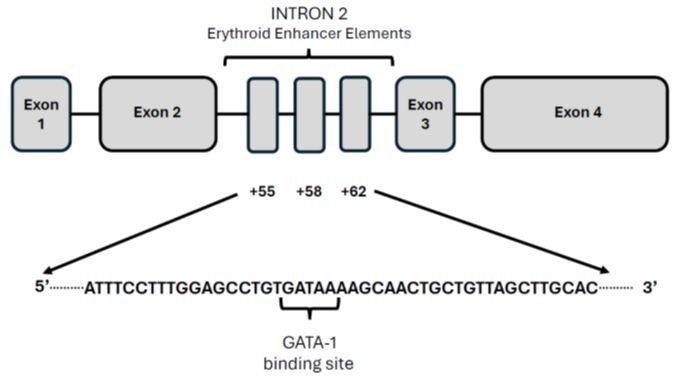
The expression of BCL11A in erythroid cells is controlled by an erythroid-specific enhancer located at position +58 in intron 2 of the BCL11A gene.[7] The +58 BCL11A intronic enhancer contains a binding site for GATA1, which is required to sustain erythroid-specific BCL11A expression. Disruption of this enhancer abolishes the expression of BCL11A, specifically in erythroid cells but not in other tissues, and does not impair the engraftment capability of HSCs.[8] Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin synthesis in erythroid cells, including those of -thal patients.[9]
BCL11A expression is transcriptionally controlled during development. In fetal erythroid cells, BCL11A expression is inhibited by the repressor HIC2, whose expression is high in fetal cells but low in adult erythroid cells.[5] In turn, HIC2 expression is controlled by some microRNAs pertaining to the let-7 miRNA family, which act as repressors of HIC2 expression.[6]
The expression of BCL11A in erythroid cells is controlled by an erythroid-specific enhancer located at position +58 in intron 2 of the BCL11A gene.[7] The +58 BCL11A intronic enhancer contains a binding site for GATA1, which is required to sustain erythroid-specific BCL11A expression. Disruption of this enhancer abolishes the expression of BCL11A, specifically in erythroid cells but not in other tissues, and does not impair the engraftment capability of HSCs.[8] Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin synthesis in erythroid cells, including those of -thal patients.[9]
Gene
Editing Therapy Studies Based on CRISPR/Cas 9 Technology
Gene editing techniques
are based on the utilization of nucleases to generate DNA double-strand
breaks into specific regions of the genome. The most used nucleases in
clinical gene therapy are zinc finger nucleases (ZFN) and clustered
regularly-interspaced short palindromic repeats and caspase (CRISPR
associated) nucleases (CRISPR/Cas).
The CRISPR-Cas9 is a two-component system that allows for targeted repairs, insertions, or deletions in specific genomic regions. The CRISPR/Cas 9 system is composed of a Cas9 that is driven on a specific DNA target by a single-guide RNA (defined as guide RNA, gRNA). The gRNA is composed of a scaffold sequence necessary for Cas-binding and a user-defined 20-nucleotide spacer that defines the specific genomic target structure to be modified. The gRNA is flanked by a DNA motif known as the photospacer adjacent motif (PAM) that is inserted in the non-target strand of the genomic DNA downstream of the target sequence: the Cas9 recognizes this interaction and cleaves the DNA. The cell attempts to repair double-stand breaks by using two different repair mechanisms: non-homologous end joining repair (NHEJ) and homology-directed repair (HDR). In the NHEJ process, the homologous and non-homologous broken ends of DNA created by Cas9 are joined. NHEJ is particularly active in G0 and G1 phases of the cell cycle and creates insertions or deletions at the cut site caused by the generation of frameshift mutations.
In hemoglobinopathies, a specific DNA site located at +58nt of the erythroid enhancer of the BCL11A gene is the most promising target site for gene editing (Figure 1).10 Deletion or mutation of this region using CRISPR/Cas9 induces an increase in expression of HbF-positive cells, particularly through editing of primary human hematopoietic progenitors and mouse transgenesis, the BCL11A erythroid enhancer was validated as a suitable target for fetal hemoglobin reinduction.[10]
A pilot clinical study provided the first initial evidence in favor of the safety and efficacy of CRISPR-Cas9 gene editing for b-thal and SCD in one b°/b° transfusion-dependent b-thal patient and one b°/bS transfusion-dependent and symptomatic patient.11 These patients represented the first patients enrolled in the CLIMB THAL-111 and CLIMB SCD-122 studies, respectively.11 This study was preceded by a preclinical evaluation of the efficiency of CRISPR-Cas9 gene editing in CD34+ cells obtained from normal subjects, showing a high efficiency (about 80% of the alleles at the BCL11A locus were modified, with no evidence of off-target editing); importantly, gene-edited CD34+ cells induced in vitro to erythroid differentiation produced 30% HbF, compared to 10% in unmodified cells.[11] After myeloablation, two patients (one -thal and one SCD) received autologous CD34+ cells edited with CRISPR-Cas9 targeting BCL11A erythroid enhancer; in both patients, HbF and total Hb levels increased rapidly after reinfusion of autologous CD34+-edited cells and with a follow-up of more than one year both patients had high levels of allelic editing in the bone marrow and blood, with a pan-cellular distribution of HbF at the level of RBCs; both the thalassemic patients become transfusion-independent, and in the patient with SCD no vaso-occlusive episodes were observed.[11]
Recently, the results of the CLIMB THAL-111 phase III trial were published; 52 patients with transfusion-dependent -thal received treatment with autologous CD34+ cells, gene-edited with CRISPR-Cas9 at the level of BCL11A erythroid enhancer (with the commercial name of Exaglamglogene, Exa-Cel); 35 of these patients were evaluable for development of transfusion independence with a sufficient follow-up.[12] 91% of the treated patients became transfusion independent, with a mean total Hb level of 13.1 g/dL and mean HbF level of 11.9 g/dL, distributed in 94% of RBCs.[12] Allelic editing at the BCL11A locus was detectable at one month after Exa-Cel infusion, and at 6 months, a mean of 78% of CD34+ bone marrow cells were genetically edited, and this percentage remained stable on time.[12] The safety profile of Exa-cel was consistent with that related to busulfan conditioning.[12]
The results reported for Exa-Cel in b-thal patients were confirmed by a similar gene-editing system based on CRISPR-Cas9-mediated disruption of the BCL11A erythroid enhancer; particularly, Fu et al initially showed in two b-thal patients the safety and the efficacy of gene editing approach based on the disruption of the GATA1-binding site at the +58 BCL11A erythroid enhancer.[13] In a more recent report, autologous, gene-edited CD34+ cells with disruption of the BCL11A erythroid enhancer (BRL-101) were reinfused in 10 b-thal patients (5 b°/b°), resulting in a sustained increase of total Hb and HbF levels, with 98-99% F-cells and achieving transfusion independence in all patients.[14] Adverse events are those associated with myeloablation and aHSCT.[14]
The results of the phase III CLIMB SCD-121 study were recently reported in a group of 44 transfusion-dependent and symptomatic SCD patients treated with Exa-Cel; 30 of these patients were evaluable for the occurrence of vaso-occlusive crises for at least 12 consecutive months.15 97% of treated patients were free from hospitalizations for severe vaso-occlusive crises for at least 12 consecutive months.[15] However, the analysis of the whole population of 44 SCD patients who received Exa-Cel infusion showed 6 events of severe vaso-occlusive crises occurring in patients whose increase of total Hb and HbF levels were similar to those observed in patients without severe vaso-occlusive events.[15] Total Hb levels increased from <9 g/dL to 12.5 g/dL 6 months after Exa-cel infusion, with 44.5 of HbF (present in >95% RBCs). All patients were transfusion-independent after treatment; the percentage of edited BCL11A alleles was 53.5% at one month and at least 70% from month 2 through the end of follow-up.15 All the 30 patients included in the primary efficacy populations showed a significant reduction of hemolysis.15 As for b-thal patients, adverse events were those expected for myeloablative busulfan conditioning and aHSCT.[15]
Other studies have explored a different approach based on CRISPR-Cas9 technology to create an HPFH (hereditary persistence of fetal hemoglobin) genotype inHSCs.[16] HPFH is a benign condition characterized by the maintenance of HbF synthesis in postnatal life. Particularly, CRISPR-Cas9-mediated genome editing of human blood progenitors was used to mutate 1 13-nt sequence that is present in the promoters of the g1 (HBG1) and g2 (HBG2) globin genes, recapitulating a naturally occurring HPFH-associated mutation (Figure 2).[17]
The HBG1 and HBG2 promoter elements are physiologically bound by the transcriptional repressor BCL11A, and their disruption by CRISPR-Cas9 single guide ribonucleoprotein complex in normal or SCD CD34+ cells determine the production of an erythroid progeny with a high HbF potential; no off-target mutations were detected in engineered CD34+ cells (Figure 2).[18] These observations have supported the safety, feasibility and efficacy of CRISPR-Cas9 gene editing of HBG1 and HBG2 promoters to induce robust levels of HbF synthesis, suitable for treating hemoglobinopathies.[18] These studies have provided the preclinical support for a clinical trial involving gene editing of HBG1 and HBG2 promoters using CRISPR-Cas 9 technology. To this end, Sharma et al. have first defined the gRNA targeting HBG1 and HBG2 promoters providing the optimal HbF induction in CD34+ cells obtained from healthy donors and patients with SCD; thus, the gRNA 68 was selected for clinical development. In this study, cells gene-edited with gRNA 68 were defined as OTQ923. Three patients with SCD received autologous OTQ923 after myeloablative conditioning, with a follow-up of 6 to 18 months; in patient 1 (bS/bS), before OTQ infusion, total Hb level was 10g/dL, with 0.4% of HbF and 4% of F cells and 12-18 months after infusion, total Hb ranged from 10.3 to 11.9 g/dL, with 25% to 26.8% HbF and 78% to 88% of F cells; in patient 2 (bS/bS), before OTQ923 infusion, total Hb level was 7.6 g/dL with 4.2% HbF and 20.4% F cells and 6-12 months after infusion, total Hb ranged from 10.1 to 11.5 g/dL, with 23% to 25% HbF and 80% to 87% F cells; in patient 3 (bS/bS), before OTQ923 infusion, total Hb level was 8.3 g/dL, with 1.4% HbF and 6.2% F cells and 4-6 months after treatment, total Hb level maintained at 10.5 g/dL, with 19% to 23% of HbF and 70% to 85% of F cells.[19] The three patients had several adverse events, all considered to be related to either myeloablative busulfan conditioning or to the underlying SCD. Particularly, after undergoing OTQ923 infusion, all three enrolled patients had at least one-related event (vaso-occlusive event).[19] Although all three participants had at least one episode of vaso-occlusive crisis after infusion, the frequency of these episodes was very limited and seemingly compatible with the induction levels of HbF obtained with this treatment; in fact, despite improvement of hematologic levels, all three participants had still ongoing mild hemolysis, suggesting that their levels of HbF were not sufficient to inhibit HbS polymerization completely.[19]
Another study based on the gene editing of the HBG1 and HBG2 promoters utilized CRISPR-Cas12 technology. The CRISPR-Cas12a is a programmable single gRNA endonuclease system. Some notable differences exist between Cas-12 and Cas-9: Cas 12a possesses a single nuclease domain and intrinsic RNA processing activity, allowing multigene editing of RNA transcripts and results in staggered DNA ends, promoting HDR repair instead of NHEJ.[20] The CRISPR-Cas12a system was used to obtain efficient editing of the distal CCAAT-box region of the HBG1 and HBG2 promoters, resulting in a high rate of editing (80-90%) of these sites in CD34+ cells, with a high rate of HbF synthesis (about 40%) in the erythroid progeny generated by gene-edited progenitors.21 Gene-edited HSCs efficiently repopulate multilineage hematopoiesis. Furthermore, no off-target editing was observed.[21] Two-phase I/II clinical trials, RUBY and EdiThal, evaluated the safety and the efficacy of EDIT-301, autologous CD34+ cells edited at the level of HBG1 and HBG2 gene promoters by CRISPR-Cas12a technology, in SCD and -thal patients, respectively; the results of the first 2 and 7 patients enrolled in the RUBY and EdiThal studies, respectively, were recently reported.[22] Successful engraftment was observed in all patients; in SCD patients, a rapid and sustained normalization of Hb as early as 4 months after infusion (from 10.5 g/dL at baseline to 14.2 g/dL after EDIT-301 infusion), an increase of HbF levels (at 4 months, 6.8 g/dL) and percentage of F cells, resolution of vascular occlusive events and an improvement or normalization of markers of hemolysis; in the two -thal patients, early improvements were observed after EDIT-301 infusion and development of transfusion-independence.[22]
The CRISPR-Cas9 is a two-component system that allows for targeted repairs, insertions, or deletions in specific genomic regions. The CRISPR/Cas 9 system is composed of a Cas9 that is driven on a specific DNA target by a single-guide RNA (defined as guide RNA, gRNA). The gRNA is composed of a scaffold sequence necessary for Cas-binding and a user-defined 20-nucleotide spacer that defines the specific genomic target structure to be modified. The gRNA is flanked by a DNA motif known as the photospacer adjacent motif (PAM) that is inserted in the non-target strand of the genomic DNA downstream of the target sequence: the Cas9 recognizes this interaction and cleaves the DNA. The cell attempts to repair double-stand breaks by using two different repair mechanisms: non-homologous end joining repair (NHEJ) and homology-directed repair (HDR). In the NHEJ process, the homologous and non-homologous broken ends of DNA created by Cas9 are joined. NHEJ is particularly active in G0 and G1 phases of the cell cycle and creates insertions or deletions at the cut site caused by the generation of frameshift mutations.
In hemoglobinopathies, a specific DNA site located at +58nt of the erythroid enhancer of the BCL11A gene is the most promising target site for gene editing (Figure 1).10 Deletion or mutation of this region using CRISPR/Cas9 induces an increase in expression of HbF-positive cells, particularly through editing of primary human hematopoietic progenitors and mouse transgenesis, the BCL11A erythroid enhancer was validated as a suitable target for fetal hemoglobin reinduction.[10]
 |
|
A pilot clinical study provided the first initial evidence in favor of the safety and efficacy of CRISPR-Cas9 gene editing for b-thal and SCD in one b°/b° transfusion-dependent b-thal patient and one b°/bS transfusion-dependent and symptomatic patient.11 These patients represented the first patients enrolled in the CLIMB THAL-111 and CLIMB SCD-122 studies, respectively.11 This study was preceded by a preclinical evaluation of the efficiency of CRISPR-Cas9 gene editing in CD34+ cells obtained from normal subjects, showing a high efficiency (about 80% of the alleles at the BCL11A locus were modified, with no evidence of off-target editing); importantly, gene-edited CD34+ cells induced in vitro to erythroid differentiation produced 30% HbF, compared to 10% in unmodified cells.[11] After myeloablation, two patients (one -thal and one SCD) received autologous CD34+ cells edited with CRISPR-Cas9 targeting BCL11A erythroid enhancer; in both patients, HbF and total Hb levels increased rapidly after reinfusion of autologous CD34+-edited cells and with a follow-up of more than one year both patients had high levels of allelic editing in the bone marrow and blood, with a pan-cellular distribution of HbF at the level of RBCs; both the thalassemic patients become transfusion-independent, and in the patient with SCD no vaso-occlusive episodes were observed.[11]
Recently, the results of the CLIMB THAL-111 phase III trial were published; 52 patients with transfusion-dependent -thal received treatment with autologous CD34+ cells, gene-edited with CRISPR-Cas9 at the level of BCL11A erythroid enhancer (with the commercial name of Exaglamglogene, Exa-Cel); 35 of these patients were evaluable for development of transfusion independence with a sufficient follow-up.[12] 91% of the treated patients became transfusion independent, with a mean total Hb level of 13.1 g/dL and mean HbF level of 11.9 g/dL, distributed in 94% of RBCs.[12] Allelic editing at the BCL11A locus was detectable at one month after Exa-Cel infusion, and at 6 months, a mean of 78% of CD34+ bone marrow cells were genetically edited, and this percentage remained stable on time.[12] The safety profile of Exa-cel was consistent with that related to busulfan conditioning.[12]
The results reported for Exa-Cel in b-thal patients were confirmed by a similar gene-editing system based on CRISPR-Cas9-mediated disruption of the BCL11A erythroid enhancer; particularly, Fu et al initially showed in two b-thal patients the safety and the efficacy of gene editing approach based on the disruption of the GATA1-binding site at the +58 BCL11A erythroid enhancer.[13] In a more recent report, autologous, gene-edited CD34+ cells with disruption of the BCL11A erythroid enhancer (BRL-101) were reinfused in 10 b-thal patients (5 b°/b°), resulting in a sustained increase of total Hb and HbF levels, with 98-99% F-cells and achieving transfusion independence in all patients.[14] Adverse events are those associated with myeloablation and aHSCT.[14]
The results of the phase III CLIMB SCD-121 study were recently reported in a group of 44 transfusion-dependent and symptomatic SCD patients treated with Exa-Cel; 30 of these patients were evaluable for the occurrence of vaso-occlusive crises for at least 12 consecutive months.15 97% of treated patients were free from hospitalizations for severe vaso-occlusive crises for at least 12 consecutive months.[15] However, the analysis of the whole population of 44 SCD patients who received Exa-Cel infusion showed 6 events of severe vaso-occlusive crises occurring in patients whose increase of total Hb and HbF levels were similar to those observed in patients without severe vaso-occlusive events.[15] Total Hb levels increased from <9 g/dL to 12.5 g/dL 6 months after Exa-cel infusion, with 44.5 of HbF (present in >95% RBCs). All patients were transfusion-independent after treatment; the percentage of edited BCL11A alleles was 53.5% at one month and at least 70% from month 2 through the end of follow-up.15 All the 30 patients included in the primary efficacy populations showed a significant reduction of hemolysis.15 As for b-thal patients, adverse events were those expected for myeloablative busulfan conditioning and aHSCT.[15]
Other studies have explored a different approach based on CRISPR-Cas9 technology to create an HPFH (hereditary persistence of fetal hemoglobin) genotype inHSCs.[16] HPFH is a benign condition characterized by the maintenance of HbF synthesis in postnatal life. Particularly, CRISPR-Cas9-mediated genome editing of human blood progenitors was used to mutate 1 13-nt sequence that is present in the promoters of the g1 (HBG1) and g2 (HBG2) globin genes, recapitulating a naturally occurring HPFH-associated mutation (Figure 2).[17]
|
|
The HBG1 and HBG2 promoter elements are physiologically bound by the transcriptional repressor BCL11A, and their disruption by CRISPR-Cas9 single guide ribonucleoprotein complex in normal or SCD CD34+ cells determine the production of an erythroid progeny with a high HbF potential; no off-target mutations were detected in engineered CD34+ cells (Figure 2).[18] These observations have supported the safety, feasibility and efficacy of CRISPR-Cas9 gene editing of HBG1 and HBG2 promoters to induce robust levels of HbF synthesis, suitable for treating hemoglobinopathies.[18] These studies have provided the preclinical support for a clinical trial involving gene editing of HBG1 and HBG2 promoters using CRISPR-Cas 9 technology. To this end, Sharma et al. have first defined the gRNA targeting HBG1 and HBG2 promoters providing the optimal HbF induction in CD34+ cells obtained from healthy donors and patients with SCD; thus, the gRNA 68 was selected for clinical development. In this study, cells gene-edited with gRNA 68 were defined as OTQ923. Three patients with SCD received autologous OTQ923 after myeloablative conditioning, with a follow-up of 6 to 18 months; in patient 1 (bS/bS), before OTQ infusion, total Hb level was 10g/dL, with 0.4% of HbF and 4% of F cells and 12-18 months after infusion, total Hb ranged from 10.3 to 11.9 g/dL, with 25% to 26.8% HbF and 78% to 88% of F cells; in patient 2 (bS/bS), before OTQ923 infusion, total Hb level was 7.6 g/dL with 4.2% HbF and 20.4% F cells and 6-12 months after infusion, total Hb ranged from 10.1 to 11.5 g/dL, with 23% to 25% HbF and 80% to 87% F cells; in patient 3 (bS/bS), before OTQ923 infusion, total Hb level was 8.3 g/dL, with 1.4% HbF and 6.2% F cells and 4-6 months after treatment, total Hb level maintained at 10.5 g/dL, with 19% to 23% of HbF and 70% to 85% of F cells.[19] The three patients had several adverse events, all considered to be related to either myeloablative busulfan conditioning or to the underlying SCD. Particularly, after undergoing OTQ923 infusion, all three enrolled patients had at least one-related event (vaso-occlusive event).[19] Although all three participants had at least one episode of vaso-occlusive crisis after infusion, the frequency of these episodes was very limited and seemingly compatible with the induction levels of HbF obtained with this treatment; in fact, despite improvement of hematologic levels, all three participants had still ongoing mild hemolysis, suggesting that their levels of HbF were not sufficient to inhibit HbS polymerization completely.[19]
Another study based on the gene editing of the HBG1 and HBG2 promoters utilized CRISPR-Cas12 technology. The CRISPR-Cas12a is a programmable single gRNA endonuclease system. Some notable differences exist between Cas-12 and Cas-9: Cas 12a possesses a single nuclease domain and intrinsic RNA processing activity, allowing multigene editing of RNA transcripts and results in staggered DNA ends, promoting HDR repair instead of NHEJ.[20] The CRISPR-Cas12a system was used to obtain efficient editing of the distal CCAAT-box region of the HBG1 and HBG2 promoters, resulting in a high rate of editing (80-90%) of these sites in CD34+ cells, with a high rate of HbF synthesis (about 40%) in the erythroid progeny generated by gene-edited progenitors.21 Gene-edited HSCs efficiently repopulate multilineage hematopoiesis. Furthermore, no off-target editing was observed.[21] Two-phase I/II clinical trials, RUBY and EdiThal, evaluated the safety and the efficacy of EDIT-301, autologous CD34+ cells edited at the level of HBG1 and HBG2 gene promoters by CRISPR-Cas12a technology, in SCD and -thal patients, respectively; the results of the first 2 and 7 patients enrolled in the RUBY and EdiThal studies, respectively, were recently reported.[22] Successful engraftment was observed in all patients; in SCD patients, a rapid and sustained normalization of Hb as early as 4 months after infusion (from 10.5 g/dL at baseline to 14.2 g/dL after EDIT-301 infusion), an increase of HbF levels (at 4 months, 6.8 g/dL) and percentage of F cells, resolution of vascular occlusive events and an improvement or normalization of markers of hemolysis; in the two -thal patients, early improvements were observed after EDIT-301 infusion and development of transfusion-independence.[22]
Post-Transcriptional Genetic Silencing of BCL11A
It is important to note
that in addition to gene editing studies, another approach of gene
therapy targeting BCL11A was based on the use of a lentiviral vector
encoding a short hairpin RNA targeting BCL11A mRNA embedded in a
microRNA, allowing erythroid-specific knockdown of BCL11A.[23] This study
enrolled six SCD patients who received an infusion of autologous CD34+
cells with the BCH-BB694 lentiviral vector post-transcriptionally
silencing BCL11A; all these patients had engraftment and achieved
robust and stable HbF synthesis (ranging from 20% to 41%) with a wide
cellular distribution (F cells ranging from 59% to 93%) and HbF per F
cells ranging from 9 to 18.6 pg per cell.23 Clinical manifestations of
SCD were reduced or absent during the follow-up period.[23] These six
patients were explored by whole-genome sequencing at the level of HSCs
pre-gene therapy and post-gene therapy to evaluate the somatic mutation
profile and clonal landscape of genetically-modified HSCs.[24] No clonal
expansions were identified among gene-modified or unmodified cells;
however, an increased frequency of potential driver mutations
associated with clonal hematopoiesis or with myeloid neoplasms (DNMT3A
and EZH2-mutated clones) was observed in both genetically modified and
unmodified cells, thus suggesting a positive selection of mutant clones
during gene therapy.[24] This observation supports an enhanced fitness of
some HSCs harboring pre-existing driver mutations.[24]
In line with these observations, Lee et al. in their evaluation of the impact of CRSIP/HDR editing versus lentiviral transduction on long-term engraftment and clonal dynamics of HSPCs in Rhesus Macacus, observed that the long-term clonality of hematopoiesis generated by CRISPR/HDR-edited cells was oligoclonal in contrast to highly polyclonal hematopoiesis generated by lentiviral-transduced HSCs.[25]
In line with these observations, Lee et al. in their evaluation of the impact of CRSIP/HDR editing versus lentiviral transduction on long-term engraftment and clonal dynamics of HSPCs in Rhesus Macacus, observed that the long-term clonality of hematopoiesis generated by CRISPR/HDR-edited cells was oligoclonal in contrast to highly polyclonal hematopoiesis generated by lentiviral-transduced HSCs.[25]
Gene Editing Using Zinc-Finger Nucleases (ZNF)
ZNFs are chimeric
nucleases made of zinc finger protein motifs, each able to recognize a
short DNA sequence, put together to increase their DNA recognition
ability; these DNA-binding modules are linked to a non-specific DNA
cleavage domain of the Fok I endonuclease. Cleavage activity is induced
when two ZNFs recognize their target sequence, forming a catalytically
active heterodimer complex.
Smith and coworkers have reported the development of a ZNF-mediated gene editing system for disruption of the GATA-binding region of the intronic erythroid-specific BCL11A enhancer in human HSCs.[26] Based on these observations, Sangamo Therapeutics launched the PRECIZN-1 phase I/II clinical trial involving the study of the safety and efficacy of autologous HSCs gene-edited at the level of the erythroid-specific BCL11A enhancer using ZNFs (BIVVoo3).[27,28] A consistent increase in therapeutic levels of HbF was observed in three out of four patients receiving the therapy (from 12% to 41%, with levels >10 pg/RBC); three patients had no recurrence of severe vaso-occlusive crises after infusion.27,28 In one of the four treated patients, high HbF levels were not maintained in the time, and recurrence of vaso-occlusive crises was observed.[27,28]
Smith and coworkers have reported the development of a ZNF-mediated gene editing system for disruption of the GATA-binding region of the intronic erythroid-specific BCL11A enhancer in human HSCs.[26] Based on these observations, Sangamo Therapeutics launched the PRECIZN-1 phase I/II clinical trial involving the study of the safety and efficacy of autologous HSCs gene-edited at the level of the erythroid-specific BCL11A enhancer using ZNFs (BIVVoo3).[27,28] A consistent increase in therapeutic levels of HbF was observed in three out of four patients receiving the therapy (from 12% to 41%, with levels >10 pg/RBC); three patients had no recurrence of severe vaso-occlusive crises after infusion.27,28 In one of the four treated patients, high HbF levels were not maintained in the time, and recurrence of vaso-occlusive crises was observed.[27,28]
Base Editing
In recent years,
significant progress has been made in the development of gene-editing
techniques based on Base Editors (BEs), which can convert specific base
pairs into desired sequences without creating DNA strand breaks and
without depending on HDR-mediated correction based on a donor DNA
template. Base editing offers various opportunities for treating SCD
through gene editing: (i) correction of the mutation present in the
-globin gene;[29] (ii) increasing HbF levels by editing transcriptional
repressors (BCL11A) or binding regions of transcriptional repressors
(HBG1 and HBG2 gene promoters); (iii) increasing HbF levels by creating
binding sites for transcriptional activators (GATA1, KLF1 and TAL1).[30]
Two clinical trials based on base editing have been launched by Beam Therapeutics, both involving SCD patients. BEAM-101 is the clinical product represented by autologous HSCs base-edited by targeting HBG1 and HBG2 promoters to activate HbF synthesis, evaluated in the context of the BEACON phase I/II study; preclinical studies have shown a high efficiency of base editing of HBG1 and HBG2 promoters using the procedure developed by Beam Therapeutics, with a high efficiency of chimerism after transplantation in immunodeficient animals and >65 of HbF synthesis in erythroid cells progeny; the first patients enrolled in this study were treated and the Beam Therapeutics developed a closed and automated process for manufacturing BEAM-101. A second study, BEAM-102, is based on a preclinical study based on the use of an engineered adenine base editor achieving efficient base editing in CD34 bS/bS cells, leading to >70% of biallelic conversion on bS to bMakassar allele and 20% monoallelic conversion of bS to bMakassar allele in vitro erythroid differentiated progeny derived from ex vivo edited CD34+ cells (the Makassar variant is naturally occurring in individuals in Southeast Asia with normal hematological parameters) and is yet to be launched.[31]
It is important to note that base editing cannot be used to create a transversion mutation necessary to correct the SCD mutation 20A>T (Glu6VAL). However, Adenine Base Editor cannot be applied to replace the A.T. with a G.C., thus generating the benign anti-sickling Hb-Makassar variant.
A base editing strategy was developed for the correction of the most prevalent -thal mutations (IVS1-110[G>A]) using the SpRY-ABE8e base editor; RNA delivery of the base editing system was safe and led to about 80% of gene correction in HSCs of patients with -thal without causing dangerous double-strand DNA breaks.32 In gene-edited HPC-derived erythroid cells, this strategy was able to restore -globin production and dyserythropoiesis observed in b-thal.[32] An alternative approach of base editing of ISV b-thal consisted of the use of ABEs with relaxed PAM requirements, with the specific aim of correcting the ISV-110 splice defect in patient-derived CD34+ cells by altering an upstream sequence element critical for aberrant splicing.[33]
A recent study reported the efficient base editing of HBG1 and HBG2 promoters using transformer base editor (tBE), a cytosine base editor with no detectable off-target mutations; disruption with tBE of the six motifs present in HBG1 and HBG2 promoters resulted in the highest g-globin expression, higher than that observed using Cas9 nuclease or conventional B.E.s.[34] In experimental models, durable therapeutic editing by tBE persisted in HSCs, supporting its potential for the treatment of hemoglobinopathies.[34]
Two clinical trials based on base editing have been launched by Beam Therapeutics, both involving SCD patients. BEAM-101 is the clinical product represented by autologous HSCs base-edited by targeting HBG1 and HBG2 promoters to activate HbF synthesis, evaluated in the context of the BEACON phase I/II study; preclinical studies have shown a high efficiency of base editing of HBG1 and HBG2 promoters using the procedure developed by Beam Therapeutics, with a high efficiency of chimerism after transplantation in immunodeficient animals and >65 of HbF synthesis in erythroid cells progeny; the first patients enrolled in this study were treated and the Beam Therapeutics developed a closed and automated process for manufacturing BEAM-101. A second study, BEAM-102, is based on a preclinical study based on the use of an engineered adenine base editor achieving efficient base editing in CD34 bS/bS cells, leading to >70% of biallelic conversion on bS to bMakassar allele and 20% monoallelic conversion of bS to bMakassar allele in vitro erythroid differentiated progeny derived from ex vivo edited CD34+ cells (the Makassar variant is naturally occurring in individuals in Southeast Asia with normal hematological parameters) and is yet to be launched.[31]
It is important to note that base editing cannot be used to create a transversion mutation necessary to correct the SCD mutation 20A>T (Glu6VAL). However, Adenine Base Editor cannot be applied to replace the A.T. with a G.C., thus generating the benign anti-sickling Hb-Makassar variant.
A base editing strategy was developed for the correction of the most prevalent -thal mutations (IVS1-110[G>A]) using the SpRY-ABE8e base editor; RNA delivery of the base editing system was safe and led to about 80% of gene correction in HSCs of patients with -thal without causing dangerous double-strand DNA breaks.32 In gene-edited HPC-derived erythroid cells, this strategy was able to restore -globin production and dyserythropoiesis observed in b-thal.[32] An alternative approach of base editing of ISV b-thal consisted of the use of ABEs with relaxed PAM requirements, with the specific aim of correcting the ISV-110 splice defect in patient-derived CD34+ cells by altering an upstream sequence element critical for aberrant splicing.[33]
A recent study reported the efficient base editing of HBG1 and HBG2 promoters using transformer base editor (tBE), a cytosine base editor with no detectable off-target mutations; disruption with tBE of the six motifs present in HBG1 and HBG2 promoters resulted in the highest g-globin expression, higher than that observed using Cas9 nuclease or conventional B.E.s.[34] In experimental models, durable therapeutic editing by tBE persisted in HSCs, supporting its potential for the treatment of hemoglobinopathies.[34]
Prime Editing
Prime editing is a gene
editing technology of recent development, conceived to bypass the
limitations of other gene editing technologies. This system is based on
some constituents and, particularly, on a PE2 component, a Maloney
leukemia virus reverse transcriptase fused to the C terminus of Cas9
nuclease, to generate a single strand break (SSB) three bases upstream
of the PAM site in this system, the gRNA is called pegRNA, and is made
by the classical gRNA directing all the editing machinery to the
specific target DNA and an edit-containing RNA template extension,
containing the information that will replace the target DNA. The
mechanism of prime editing involves an initial formation of SSB by the
PE2 component, allowing the resultant 3' flap of the nicked DNA strand
to form a sequence-specific interaction with the primer binding site on
the peg RNA.[35] In a preclinical study, Everette and coworkers showed
that prime editing could correct the HbS allele to wild-type HbA at
frequencies of 15-41% in HSPCs derived from SCD patients; prime-edited
HSPCs displayed high engraftment capacities and maintained HbA
expression and lineage maturation as well as HSPCs from healthy donors;
minimal off-targeting editing was observed at 100 potential sites.[36] In
a second study, prime editing in vivo was developed in a mouse model of
SCD (CD46/Townes mice): a single-editor expressing viral vector into
mobilized CD46/Townes mice resulted in a mean value of 43% of HbS
replacement by HbA, with a marked improvement of the SCD phenotype.[37]
Conclusions
The treatment landscape
for hemoglobinopathies gained a novel addition when the FDA and EMA
approved the CRISPR-CAS9 gene editing therapy (Exa-cel) for the
treatment of patients with SCD and transfusion-dependent -thal. This
was the first gene editing therapy to receive regulatory approval and
opened the way to a new era in the treatment of monoallelic genetic
diseases.
However, despite the very favorable results obtained in these studies and the exciting implications for patients with SCD and thal, the exorbitant cost of Exa-cel, estimated to be in the order of $2.2 million per patient, is expected to greatly limit access to this revolutionary therapy. Furthermore, the healthcare structures suitable for administering autologous gene therapy are limited.
We need to follow patients longer to evaluate the long-term efficacy and safety of gene editing-based therapies. Future studies must assess the possible consequences of off-target editing and the effects of the gene editing procedure on clonal hematopoiesis and on the expansion of hematopoietic cells bearing mutations potentially associated with myeloid malignancies.
However, despite the very favorable results obtained in these studies and the exciting implications for patients with SCD and thal, the exorbitant cost of Exa-cel, estimated to be in the order of $2.2 million per patient, is expected to greatly limit access to this revolutionary therapy. Furthermore, the healthcare structures suitable for administering autologous gene therapy are limited.
We need to follow patients longer to evaluate the long-term efficacy and safety of gene editing-based therapies. Future studies must assess the possible consequences of off-target editing and the effects of the gene editing procedure on clonal hematopoiesis and on the expansion of hematopoietic cells bearing mutations potentially associated with myeloid malignancies.
References
- Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell A, Fisher C, Suclu M, Martyn G, Norton LJ. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016; 351: 285-289. https://doi.org/10.1126/science.aad3312 PMid:26816381 PMCid:PMC4778394
- Lidonnici MR, Scaramuzza S, Ferrari G. Gene therapy for hemoglobinopathies. Gene Therapy 2023; 34: 793-807. https://doi.org/10.1089/hum.2023.138 PMid:37675899
- Locatelli F, Thompson AA, Kwiatowski JL, Porter JB, Thraser AJ, Hongeng S, Sauer MG, Thuret I, Lai A, Thraser AJ, et al. Betibeglogene autotemcel gene therapy for non-0 /0 genotype -thalassemia. N Engl J Med 2022; 386: 415-427. https://doi.org/10.1056/NEJMoa2113206 PMid:34891223
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Marcias-Travino C, Rogers JM, Kurita R, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 2018; 173: 430-442. https://doi.org/10.1016/j.cell.2018.03.016 PMid:29606353 PMCid:PMC5889339
- Huang P, Pesiak CA, Ren R, Khandros E, Qin K, Keller CA, Giardine B, Bell HW, Lan X, Sharma M, et al. HIC2 controls developmental hemoglobin switching by repressing BCL11A transcription. Nat Genet 2022; 54: 1417-1426. https://doi.org/10.1038/s41588-022-01152-6 PMid:35941187 PMCid:PMC9940634
- Huang P, Pesiak SA, Shehu V, Keller CA, Giardine B, Shi J, Hardison RC, Blobel GA, Khandros E. Let-7 miRNAs repress HIC2 to regulate BCL11A transcription and hemoglobin switching. Blood 2024; 143: 1980-1991. https://doi.org/10.1182/blood.2023023399 PMid:38364109
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013; 342: 2453-257. https://doi.org/10.1126/science.1242088 PMid:24115442 PMCid:PMC4018826
- Smith EC, Luc S, Croney DM, Woodworth MB, Greig LC, Fujiwara Y, Nguyen M, Sher F, Macklis JD, Bauer DE, et al. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 2016; 128: 2338-2342. https://doi.org/10.1182/blood-2016-08-736249 PMid:27707736 PMCid:PMC5106112
- Psatha N, Reik A; Phelps S, Zhou Y, Dalas D, Yannaki E, Levasseur DN, Urnov FD, Holmes MC, Papayannopoulou T. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with -thalassemia major. Mol Therapy Methods & Clin Dev 2018; 10: 313-320. https://doi.org/10.1016/j.omtm.2018.08.003 PMid:30182035 PMCid:PMC6120587
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; 527: 192-197. https://doi.org/10.1038/nature15521 PMid:26375006 PMCid:PMC4644101
- Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, et al. CRISPR-Casp gene editing for sickle cell disease and -thalassemia. N Engl J Med 2022; 384: 252-260. https://doi.org/10.1056/NEJMoa2031054 PMid:33283989
- Locatelli F, Lang P, Wall D, Meisel R, Carbacioglu S, Li AM, de la Fuente J, Shah AJ, Carpenter B, Kwiatkowski JL, et al. Examglogene autotemcel for transfusion-dependent -thalassemia. N Engl J Med 2024; 390: 1663-1676. https://doi.org/10.1056/NEJMoa2309673 PMid:38657265
- Fu B, Liao J, Chen S, Li W, Wang Q, Hu J, Yang F, Hsiao S, Jiang Y, Wang L, et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric °/° transfusion-dependent -thalassemia. Nat Med 2022; 28: 1573-1580. https://doi.org/10.1038/s41591-022-01906-z PMid:35922667
- Zheng B,
Liu R, Zhang X, Fu B, Xu Y, Shi J, Feng X, Wang L, Wang C, Liang R, et
al. Efficacy and safety of Brl-101, CRISPR-Cas9-mediated gene editing
of the BCL11A enhancer in transfusion-dependent -thalassemia. Blood
2023; 142 (suppl.1): 4995. https://doi.org/10.1182/blood-2023-186031
- Frangoul H, Locatelli F, Bhatia M, Mapara M, Molinari L, Wall D, Liem RI, Telfer P, Shah AJ, Cavazzana M, et al. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med 2024; 390: 1649-1662. https://doi.org/10.1056/NEJMoa2309676 PMid:38661449
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, Kan YW. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and -thalassemia. Proc Natl Acad Sci USA 2016; 113: 10661-10665. https://doi.org/10.1073/pnas.1612075113 PMid:27601644 PMCid:PMC5035856
- Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, et al. A genome-editing strategy to treat -hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016; 22: 987-990. https://doi.org/10.1038/nm.4170 PMid:27525524 PMCid:PMC5706766
- Métais JY, Doerfler PA, Mayuranathan T, Bauer DE, Fowler SC, Hsieh MM, Katta V, Kerwala S, Lazzarotto CR, Luk K, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 2019; 3: 3379-3392. https://doi.org/10.1182/bloodadvances.2019000820 PMid:31698466 PMCid:PMC6855127
- Sharma A, Boelens JJ, Cancio M, Hanking JS, Bhad P, Azizy M, Lewandowski A, Zhao X, Chitnis S, Peddinti R, et al. CRISPR-Cas9 editing of the HBBG1 and HBG2 promoters to treat sickle cell disease. N Engl J Med 2023; 389: 820-832. https://doi.org/10.1056/NEJMoa2215643 PMid:37646679
- Paul B, Montoya G. CRISPR-Cas12a: functional overview and applications. Biochem J 2020; 43: 8-17. https://doi.org/10.1016/j.bj.2019.10.005 PMid:32200959 PMCid:PMC7090318
- De Dreuzy E, Haeth J, Zuris JA, Sousa P, Viswanathan R, Scott S, Da Silva J, Ta T, Copehart S, Wang T, et al. EDIT-301: an experimental autologous cell therapy comprising Cas12a-RNP modified mPB-CD34+ cells for the potential treatment of SCD. Blood 2019; 134(suppl.1): 4636. https://doi.org/10.1182/blood-2019-130256
- Hanna R, Frangoul H, McKinney C, Pineiro L, Mapara M, Chang KH, Jaskolka M, Kim K, Farrington DL, Wally M, et al. AsCas12a gene editing of HBG1/2 promoters with EDIT-301 results in rapid and sustained normalization of hemoglobin and increased fetal hemoglobin in patients with severe sickle cell disease and transfusion-dependent beta-thalassemia. Blood 2023; 142(suppl.1): 4996. https://doi.org/10.1182/blood-2023-187397
- Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, Daley H, MacKinnon B, Morris E, Federico A, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med 2021; 384: 205-215. https://doi.org/10.1056/NEJMoa2029392 PMid:33283990 PMCid:PMC7962145
- Spencer-Chapman M, Cull AH, Ciuculescu MF, Esrick EB, Mitchell E, Jung H, O'Neill L, Roberts K, Fabre MA, et al. Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease. Nat Med 2023; 29: 3175-3183. https://doi.org/10.1038/s41591-023-02636-6 PMid:37973947 PMCid:PMC10719109
- Lee BC, Gin A, Wu C, Singh K, Grice M, Mortlock R, Abraham D, Fan X, Zhou Y, AIJanahi A, et al. Impact of CRISPR/HDR editing versus lentiviral transduction on long-term engraftment and clonal dynamics of HSCPCs in rhesus macaques. Cell Stem Cell 2024; 31: 455-466. https://doi.org/10.1016/j.stem.2024.02.010 PMid:38508195
- Smith AR, Schiller GJ, Vercellotti GM, Kwiatowski JL, Krishnamurti L, Esrick EB, Williams DA, Miller WP, Woolfson A; Walters MC. Preliminary results of a phase 1 / 2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood 2019; 134(suppl.1): 3544. https://doi.org/10.1182/blood-2019-125743
- Alavi A, Krishnamurti L, Abedi M, Galeon I, Reiner D, Smith SE, Wang L, Ramezi A, Rendo P, Walters MC. Preliminary safety and efficacy results from Precizn-1: an ongoing phase 1 / 2 study on zinc finger nuclease-modified autologous CD34+ HSPCs for sickle cell disease (SCD). Blood 2021; 138(suppl.1): Blood 2021; 138(suppl.1): 2930. https://doi.org/10.1182/blood-2021-151650
- Alavi A, Abedi M, Parikh S, Boismenu R, Chen M, Hsu BL, Cockroft BM, Galeon I, Rendo P, Walters MC. Inetrin safety and efficacy results from a phase 1 / 2 study of zinc finger nuclease-modified autologous hematopoietic stem cells for sickle cell disease (PRECIZN-1). Blood 2022; 140(suppl.1): 4907-4909. https://doi.org/10.1182/blood-2022-163725
- Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, Sheppard-Tillman H, Porter SN, Yao Y, Mayberry K, et al. Base editing of hematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595: 295-302. https://doi.org/10.1038/s41586-021-03609-w PMid:34079130 PMCid:PMC8266759
- Mayuranathan T, Newby GA, Feng R, Yao Y, Mayberry KD, Lazzarotto CR, Li Y, Levine RM, Nimmagadda N, Dempsey E, et al. Potent and uniform fetal hemoglobin induction via base editing. Nat Genet 2023; 55: 1210-1220. https://doi.org/10.1038/s41588-023-01434-7 PMid:37400614 PMCid:PMC10722557
- Chu SH, Ortega M, Feliciano P, Winton V, Xu C, Haupt D, McDonald T, Martinez S, Liquori A, Marshall J, et al. Conversion of HbS to Nb G-Makassar by adenine base editing is compatible with normal hemoglobin function. Blood 2021; 138(suppl.1): 951-952. https://doi.org/10.1182/blood-2021-150922
- Hardouin G, Anatoniou P, Martinucci P, Felix T, Manceau S, Joseph L, Masson C, Scaramuzza S, Ferrari G, Cavazzana M, et al. Adenine base editor-mediated correction of the common and severe IVS1-110 (G>A) -thalassemia mutation. Blood 2023; 141: 1169-1177. https://doi.org/10.1182/blood.2022016629 PMid:36508706 PMCid:PMC10651780
- Naiiseh B, Papasavva PL, Papaioannu NY, Tomazou M, Koniali L, Felekis X, Constantinou C, Sitarou M, Christou S, Kleanthous M, et al. Context base editing for splice correction of IVSI-110 -thalassemia. Molecular Therapy Nucleic Acids 2024; 35: 1-12. https://doi.org/10.1016/j.omtn.2024.102183 PMid:38706633 PMCid:PMC11068610
- Han W, Qiu HY, Sun S, Fu ZC, Wang GQ, Qian X, Wang L, Zhai X, Wei J, Wang Y, et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs. Cell Stem Cell 2023; 30: 1624-1639. https://doi.org/10.1016/j.stem.2023.10.007 PMid:37989316
- Anzalone AW, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby JA, Raguram A, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019; 576: 149-157. https://doi.org/10.1038/s41586-019-1711-4 PMid:31634902 PMCid:PMC6907074
- Everette KA, Newby GA, Levine RM, Meyberry K, Jang Y, Jang Y, Mayuranathan T, Nimmagadda N, Dempsey E, Li Y, et al. Ex vivo prime editing of patient hematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng 2023; 7: 616-628. https://doi.org/10.1038/s41551-023-01026-0 PMid:37069266 PMCid:PMC10195679
- Li C, Georgakopoulou A, Newby GA, Chen PJ, Everette KA, Paschoudi K, Viachaki E, Gil S, Anderson AK, Koob T, et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood 2023; 141: 2085-2089. https://doi.org/10.1182/blood.2022018252 PMid:36800642 PMCid:PMC10163316