Here, we reported a Chinese female with Hb H disease who was homozygous for the αT-Saudiα mutation as a result of UPD. We explored a possible mechanism for its generation as it displayed a relatively complex structure with uniparental maternal heterodisomy and isodisomy. Moreover, while the αT-Saudiα mutation is a well-known ethnic-specific variant in the Arabian population,[5] this is the first time it has been detected in the Chinese population.
The patient (II-4) was a pregnant 25-year-old woman who lived in Guangdong Province in South China, a region of high thalassemia prevalence.6 However, she was born in Shanxi Province in northwest China, a region where thalassemia is not endemic. During the obstetrical examination process, she was diagnosed with Hb H disease on the basis of moderate anemia with microcytosis and hypochromia, a reduced HbA2 level, and the existence of the presence of Hb H and Hb Bart’s in peripheral blood cells (Table 1). She had no symptoms of splenomegaly, hepatomegaly, jaundice or iron deficiency, and she did not receive any blood transfusions. A routine test for common mutations associated with α-thalassemia in a local hospital in Guangdong showed negative results. Her family (Figure 1A) was referred to our laboratory for further analysis. Written informed consent was obtained in accordance with the Declaration of Helsinki. A detailed family history was collected, and the pure Chinese origin of the family was confirmed at least 5 generations back, with no record of migration marriage.
 |
|
 |
|
The hematological data from this family are shown in Table 1. The patient’s mother and three siblings all presented microcytic hypochromic parameters with normal hemoglobin levels and reduced or borderline Hb A2 levels, consistent with the clinical characteristics of the α-thalassemia trait, while the father showed a normal phenotype. Sanger sequencing (Figure 1B and Supplementary Figure 1) revealed the presence of the αT-Saudiα mutation (HBA2: c.*94A>G) in this family, including a homozygous in the patient and a heterozygous result in her mother and three siblings, whereas a normal result in her father. αT-Saudiα is a non-deletional α-thalassemia mutation occurring in the poly-A signal of the α2-globin gene (AATAAA>AATAAG). Previous research[5] demonstrated that homozygotes for this mutation present with typical Hb H disease (Hb ranges: 74–97 g/L and Hb H level:7.5%-27.2%). Therefore, the α-globin genotype data of all family members are in accordance with their phenotypes. Puzzlingly, however, the patient is a homozygote, while her father does not carry this mutation. Multiplex ligation-dependent probe amplification (MLPA) analysis excluded the existence of deletions (data not shown). Therefore, we speculated that the homozygous state might have been caused by UPD. Genome-wide single nucleotide polymorphism (SNP) genotyping analysis was performed using the Illumina Human Omni Zhonghua-8 BeadChip in the proband and her parents. UPD analysis of the patient was carried out using genotype data in a trio. The results (Figure 1C) showed that there were two maternal heterodisomic regions and two maternal isodisomic regions on chromosome 16. Therefore, we confirm that this case of Hb H disease was caused by maternal UPD.
To date, a total of 12 cases of hemoglobinopathy caused by UPD have been reported (Supplementary Table 1). Seven cases, comprising six cases of thalassemia major and one case of abnormal hemoglobin, were due to UPD on chromosome 11 encompassing the β-globin gene. Five cases, comprising three cases of Hb Barts hydrops foetalis, one case of Hb H disease (our case) and one case of abnormal hemoglobin, were due to UPD on chromosome 16 encompassing the α-globin gene.[7-10] When comparing our data with previously published data, we inferred that our case is only one with iUPD at the distal p (short arm) and distal q (long arm) segment and hUPD at the pericentromeric region. In addition, among the 5 cases of UPD related to α-globin, 2 of them mentioned that possible mechanism leading to UPD is nondisjunction error at maternal meiosis I. Therefore, we further investigated the mechanism of UPD formation in our case.
Mechanisms that result in UPD include trisomy rescue, gamete complementation, monosomy rescue and somatic recombination.[11] Monosomy rescue leads to complete isodisomy, and gamete complementation produces complete heterodisomy. Considering that our case has partial heterodisomy and partial isodisomy, the above two mechanisms can be ruled out. Somatic recombination always results in mosaicism. Analysis of DNA from our patient’s hair follicle and oral mucosal cell DNA was consistent with the testing of her blood cells, which excludes the existence of mosaicism in somatic cells and indicates that trisomy rescue is the only possible mechanism.
Moreover, our case had heterodisomic and isodisomic regions, suggesting the occurrence of homologous recombination events at multiple sites and segregation error events in meiosis.12 Nondisjunction error can occur in both meiosis I and meiosis II. Based on this situation, recombination occurs at distal short arm and distal long arm fragments if nondisjunction takes place in meiosis I. In contrast, if nondisjunction takes place in meiosis II, recombination occurs at pericentromeric fragments. Since the pericentromeric region is often suppressed for recombination, nondisjunction is very unlikely to occur in meiosis II. Therefore, we propose that nondisjunction can occur during meiosis I and then due to trisomy rescue, resulting in uniparental disomy, as shown in Figure 1D.
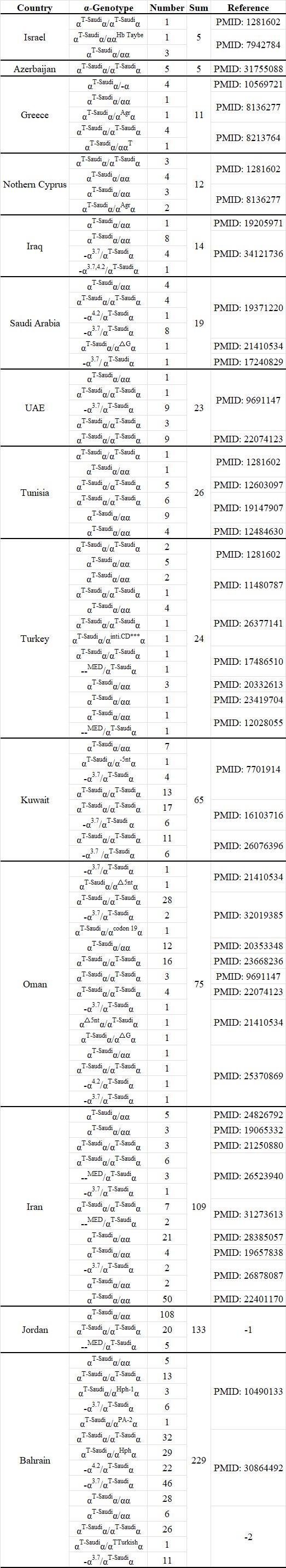
In previously published data, all known individuals carrying this mutation (Supplementary Table 2) were distributed either in countries in the Middle East or in countries with a large Arabian population (Figure 2). Previous studies have confirmed that the αT-Saudiα mutation is one of the major α-thalassemia determinants in the population of the Arabian Peninsula.5 Therefore, this mutation can be regarded as an ethnic-specific variant in the Arabian population. However, it is the first identification of the αT-Saudiα mutation in the Chinese population, and we speculated that it is population migration that results in ethnic-specific mutations spread to other populations.
 |
|
In summary, we have identified the detailed UPD pattern in this patient with Hb H disease and have analyzed the mechanism by which it originated. These data may be valuable for providing accurate diagnoses in similar cases. In addition, this case could be used as new evidence substantiating the gene flow between the Chinese population and another population in ancient times.
List of abbreviations and HGVS name of the variants
UPD, uniparental disomy; iUPD, uniparental isodisomy; hUPD, uniparental heterodisomy; SNP, single nucleotide polymorphism; MLPA, multiplex ligation-dependent probe amplification; NDJ, nondisjunction; Hb, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin. --MED: including --MED Ⅰ (NG_000006.1:g.24664_41064del16401) and –MED Ⅱ (NG_000006.1:g.10864_40864del30001).The reference sequence of HBA2 is from NC_000016.10, GRCh38.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Zhuhai Women and Children's Hospital (number IRB-2020081401). We certify that the study was performed in accordance with the 1964 Declaration of Helsinki and later amendments. Written informed consent was obtained from all the participants prior to enrollment in this study.Consent for publication
Written informed consent was obtained from the patient and her parents for publication of this Case report and any accompanying images.Availability of data and materials
The data in the current study are available within the article and its supplementary materials. The variant has been submitted to ClinVar under Accession: VCV000375749.61 (https://www.ncbi.nlm.nih.gov/clinvar/variation/375749/?oq=HBA2:%20c.*94A%3EG&m=NM_000517.6(HBA2):c.*94A%3EG)Funding
This work was supported by National Natural Science Foundation of China (82370122), Guangdong Basic and Applied Basic Research Foundation (2024A1515012748) and open foundation from Guangxi Key Laboratory of Precision Medicine for Genetic Diseases of Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region(GXWCH-ZDKF-2023–11).CRediT authorship contribution statement
Ge Wang, Yuqiu Zhou: Resources. Hongtiong Xie: Conceptualization, Data curation, Methodology, Writing – original draft. Jun Zhang, Peng Huang, Min Liang: Formal analysis, DinaZhu,Qianqian Zhang: Validation. Xuan Shang: Conceptualization, Writing – review and editing.Acknowledgements
We thank the patient and her family for their reliance and cooperation.References
- Angastiniotis M, Eleftheriou A, Galanello R, Harteveld CL, Petrou M, Traeger-Synodinos J, et al. In: Old J, editor. Prevention of Thalassaemias and Other Haemoglobin Disorders: Volume 1: Principles. Nicosia (Cyprus): Thalassaemia International Federation © 2013 Thalassaemia International Federation.; 2013.
- Shang X, Xu X. Update in the genetics of thalassemia: What clinicians need to know. Best Pract Res Clin Obstet Gynaecol. 2017;39:3-15. https://doi.org/10.1016/j.bpobgyn.2016.10.012 PMid:27876354
- Zhu C, Yu W, Xie J, Chen L, Ding H, Shang X, et al. Hemoglobin H disease due to a de novo mutation at the α2-globin gene and an inherited common α-thalassemia deletion found in a Chinese boy. Blood Cells Mol Dis. 2010;45(3):223-6. https://doi.org/10.1016/j.bcmd.2010.07.005 PMid:20691621
- Nakka P, Pattillo Smith S, O'Donnell-Luria AH, McManus KF, Mountain JL, Ramachandran S, et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am J Hum Genet. 2019;105(5):921-32. https://doi.org/10.1016/j.ajhg.2019.09.016 PMid:31607426 PMCid:PMC6848996
- Al Moamen NJ, Thabet A, Mahdi F, Newton H, Salman E. Various α-Thalassemia Genotype Combinations of the Saudi-Type Polyadenylation Signal Mutation (αT-Saudiα) in the Population of Bahrain: An Update of Genotype-Phenotype Analyses. Hemoglobin. 2018;42(3):166-70. https://doi.org/10.1080/03630269.2018.1499523 PMid:30864492
- Shang X, Peng Z, Ye Y, Asan, Zhang X, Chen Y, et al. Rapid Targeted Next-Generation Sequencing Platform for Molecular Screening and Clinical Genotyping in Subjects with Hemoglobinopathies. EBioMedicine. 2017;23:150-9. https://doi.org/10.1016/j.ebiom.2017.08.015 PMid:28865746 PMCid:PMC5605365
- Au PK, Kan AS, Tang MH, Leung KY, Chan KY, Tang TW, et al. A Fetus with Hb Bart's Disease Due to Maternal Uniparental Disomy for Chromosome 16. Hemoglobin. 2016;40(1):66-9. https://doi.org/10.3109/03630269.2015.1096283 PMid:26574185
- Tan YR, Tan HK. A Rare Case of Hemoglobin Bart's Hydrops Fetalis due to Uniparental Disomy of Chromosome 16. J Med Cases. 2021;12(7):275-9. https://doi.org/10.14740/jmc3693 PMid:34434471 PMCid:PMC8383697
- Wattanasirichaigoon D, Promsonthi P, Chuansumrit A, Leopairut J, Yanatatsaneejit P, Rattanatanyong P, et al. Maternal uniparental disomy of chromosome 16 resulting in hemoglobin Bart's hydrops fetalis. Clin Genet. 2008;74(3):284-7. https://doi.org/10.1111/j.1399-0004.2008.01046.x PMid:18564363
- Mehta N, Johnston JM, Hein M, Kipp BR, Coon L, Savedra ME, et al. Further Characterization of Hb Bronovo [α103(G10)His→Leu; HBA2: c.311A>T] and First Report of the Homozygous State. Hemoglobin. 2020;44(3):174-8. https://doi.org/10.1080/03630269.2020.1776322 PMid:32552204
- Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol Cell. 2011;103(7):303-17. https://doi.org/10.1042/BC20110013 PMid:21651501
- Benn P.
Uniparental disomy: Origin, frequency, and clinical significance.
Prenat Diagn. 2021;41(5):564-72. https://doi.org/10.1002/pd.5837
PMid:33179335
Supplementary Files
|
|
|
|
 |
|